Дефицит лизосомной кислой липазы
![]() Версия: Клинические рекомендации РФ 2023 (Россия)
Версия: Клинические рекомендации РФ 2023 (Россия)
Общая информация
Краткое описание
- Ассоциация медицинских генетиков
- Союз педиатров России
- Российская Гастроэнтерологическая Ассоциация
- Российское трансплантологическое общество
Одобрено Научно-практическим Советом Минздрава РФ
В соответствии с Правилами поэтапного перехода медицинских организаций к оказанию медицинской помощи на основе клинических рекомендаций, разработанных и утвержденных в соответствии с частями 3, 4, 6 –9 и 11 статьи 37 Федерального закона «Об основах охраны здоровья граждан в Российской Федерации», утвержденных постановлением Правительства Российской Федерации от 19.11.2021 № 1968, клинические рекомендации применяются следующим образом:
– размещенные в Рубрикаторе после 1 июня 2022 года – с 1 января 2024 года.
Возрастная категория: Взрослые, Дети
Пересмотр не позднее: 2025
Статус: Действует
Определение заболевания или состояния (группы заболеваний или состояний)
Дефицит лизосомной кислой липазы (ДЛКЛ) — прогрессирующее наследственное заболевание, в основе которого лежит дефект гена LIPA, кодирующего фермент лизосомную кислую липазу (ЛКЛ), приводящий к накоплению эфиров холестерина и триглицеридов в органах и тканях [1-3].
Особенности кодирования заболевания или состояния (группы заболеваний или состояний) по Международной статической класификации болезней и проблем, связанных со здоровьем
Согласно МКБ10, заболевание относится к классу IV, болезням эндокринной системы, расстройству питания и нарушению обмена веществ, Е75.5 Другие нарушения накопления липидов.
Классификация
Классификация заболевания или состояния (группы заболеваний или состояний)
Выделяют две основные формы ДЛКЛ (приложение А3):
- инфантильная (болезнь Вольмана) — с манифестацией в первые 6 месяцев жизни
- болезнь накопления эфиров холестерина (БНЭХ), с дебютом в возрасте старше 6 месяцев (наиболее часто в 2–5 лет) [3,46].
Следует отметить, что ДЛКЛ — это спектр клинических фенотипов с разным характером течения и прогнозом болезни [3].
Этиология и патогенез
Этиология и патогенез заболевания или состояния (группы заболеваний или состояний)
ДЛКЛ наследуется по аутосомно-рецессивному типу. Заболевание связано с мутациями гена LIPA, который картирован на 10 хромосоме (10q23.31). В международной базе данных по мутациям человека описано около 100 мутаций в гене LIPA, приводящих к нарушению функции фермента. При ДЛКЛ нонсенс-мутации, крупные перестройки гена, мутации со сдвигом рамки считывания, в гомозиготном или компаунд-гетерозиготном состоянии, как правило, выявляют при тяжелых формах заболевания [1,3,4]. Наиболее распространенный патогенный аллель — синонимичная замена, нарушающая сайт сплайсинга в экзоне 8 c.894G> A (E8SJM-1G> A), является причиной заболевания в более чем половине опубликованных случаев. Наличие данного варианта в гомозиготном или компаунд-гетерозиготном состоянии сохраняет 3–5% остаточной активности ЛКЛ [1 – 3].
В норме попавшие в лизосому путем рецептор-опосредованного эндоцитоза нейтральные жиры (эфиры холестерина и, в меньшей степени, триглицериды) под воздействием ЛКЛ расщепляются до свободного холестерина и жирных кислот. Эти липиды и их окисленные производные вступают во взаимодействие с факторами транскрипции (стериновым регуляторным элементом связывания белков — СРЭСБ), которые непосредственно модулируют экспрессию генов, вовлеченных в синтез и захват холестерина, а также липогенез. При отсутствии или снижении активности ЛКЛ эфиры холестерина, триглицериды не распадаются и накапливаются в лизосомах. Соответствующий недостаток свободного холестерина в клетке приводит к СРЭСБ-опосредованной стимуляции эндогенного синтеза холестерина ингибированию гидроксиметилглутарил-коэнзим А (ГМГ-КоА) редуктазой и эндоцитоза посредством рецепторов ЛПНП. Параллельно с этим увеличивается синтез аполипопротеина В (АпоВ) и значительно повышается образование холестерина липопротеинов очень низкой плотности (ЛПОНП). Увеличение экспрессии ГМГ-КоА редуктазы является первичным результатом СРЭСБ-2-опосредованного внутриклеточного уменьшения холестерина, приводя к увеличению уровня свободного холестерина [1, 2].
Таким образом, накопление эфиров холестерина и триглицеридов в органах и тканях сопровождается дислипидемией: в сыворотке крови определяется повышенный уровень холестерина, ЛПНП, уровень ЛПВП соответствует норме или снижен. У части пациентов регистрируется гипертриглицеридемия.
При наиболее тяжелой, младенческой форме ДЛКЛ — болезни Вольмана активность фермента составляет менее 1% от нормы, что приводит к быстрому массивному накоплению эфиров холестерина и триглицеридов в лизосомах многих органах и тканях, в первую очередь в печени, селезенке, надпочечниках, ворсинах кишечника, костном мозге, лимфатических узлах, в макрофагах ретикуло-эндотелиальной системы, что обуславливает полисистемные проявления болезни.
При болезни накопления эфиров холестерина (БНЭХ) — фенотипе ДЛКЛ, характеризующемся более медленным прогрессированием и вариабельностью клинических проявлений — in vitro определяется остаточная активность ЛКЛ в диапазоне 1-12% от нормы [1-4].
Эпидемиология
Эпидемиология заболевания или состояния (группы заболеваний или состояний)
Распространенность ДЛКЛ вариабельна в зависимости от факторов этнической принадлежности и географического положения. Считается, что частота ДЛКЛ составляет в среднем 1:40000-1:300000 живых новорожденных [3]. Частота встречаемости младенческой формы заболевания в 2-2,5 раза ниже и составляет 1 на 100 000-500 000 живорожденных новорожденных [5-7]. Исследования по изучению частоты встречаемости ДЛКЛ в России проводились в г. Москва. Данный показатель составил примерно 1:70 000 новорожденных [8].
Клиническая картина
Cимптомы, течение
Клиническая картина заболевания или состояния (группы заболеваний или состояний)
ДЛКЛ представляет собой различные фенотипы от быстро прогрессирующей летальной младенческой формы и тяжелых детских вариантов с серьезными осложнениями, например, циррозом и печеночной недостаточностью в подростковом возрасте, до субклинических, мягких форм, проявляющихся в более старшем возрасте [3].
1. Клиническая картина при инфантильной форме ДЛКЛ (болезни Вольмана)
Инфантильная форма ДЛКЛ (болезнь Вольмана, БВ) — редкое быстро прогрессирующее заболевание, проявляющееся с первых месяцев жизни, приводящее к развитию печеночной недостаточности и летальному исходу в возрасте до 1 года [9].
Наиболее характерными признаками БВ являются гастроинтестинальные проявления, включающие рвоту, диарею и/или стеаторею. Часто выявляется увеличение объёма живота за счет вздутия кишечника, гепатомегалии и/или гепатоспленомегалии, а также потеря веса с развитием гипотрофии и, в тяжелых случаях, кахексии. Также характерны задержка роста и физического развития. Кроме того, для этих пациентов характерна интермиттирующая лихорадка, вялость, астения и гиперрефлексия [2, 9,10]. У пациентов развивается анемия и тромбоцитопения.
Особенностью БВ является увеличение и кальцификация надпочечников, встречающаяся примерно в 50% случаев [2]. По данным визуализирующих методов, увеличенные надпочечники сохраняют свою полулунную или пирамидальную форму, по всей паренхиме их определяются точечные очаги кальцификации [11]. Отсутствие кальцификатов надпочечников не исключает диагноз.
Для пациентов с БВ характерна неврологическая симптоматика, обусловленная печеночной энцефалопатией.
При БВ возникает синдром мальабсорбции, связанный с недостатком жирных кислот, повреждением слизистой оболочки кишечника и проявляющийся разной степенью выраженности диареи, стеатореи, синдрома избыточного бактериального роста и метеоризма [3, 9].
Макроскопически печень значительно увеличена, желтого цвета с жирной поверхностью среза. Гепатоциты и клетки Купфера во всех зонах дольки содержат липидные вакуоли, что микроскопически соответствует мелкокапельной жировой дистрофии. В замороженных образцах при окраске суданом черным обнаруживаются липидные вакуоли, которые при оценке в поляризованном свете имеют вид красноватых кристаллов. Портальный и перипортальный фиброз часто прогрессирует в микронодулярный цирроз печени.
При ультраструктурном исследовании в лизосомах выявляют накопления субстратов, имеющих глобулярный и кристаллический вид.
Для точной морфологической диагностики в настоящее время разработана иммуногистохическая панель антител. Обнаружение катепсина D и экспрессия мембранных лизосомальных маркеров, таких как лизосомально-ассоциированные мембранные белки 1 и 2 (LAMP 1 и 2) и лизосомальный интегральный мембранный белок 2 (LIMP 2) вокруг липидных вакуолей, подтверждает внутрилизосомальное накопление липидов. Кроме того, к гистологическим особенностям относят также накопление цероида в макрофагах [3,10 ].
Описаны случаи БВ с манифестацией во внутриутробном периоде в виде некроза надпочечников, полигидроамниона, накопления эфиров холестерина (ЭХ) в органах и тканях и микровезикулярного стеатоза печени [12]. БВ может быть причиной внутриутробной гибели плода. При аутопсии на 17-й неделе беременности продемонстрировано накопление липидов в гепатоцитах и синцитиотрофобластах, а также некроз в надпочечниковой железе плода [9].
У пациентов с БВ определяется гепатомегалия или гепатоспленомегалия, быстро развивается печеночная недостаточность с исходом в цирроз печени. В биохимическом анализе крови — выраженное повышение уровня трансаминаз, значительное увеличение билирубина, синдром холестаза. Часто встречающимся лабораторным признаком при БВ является повышение уровней лактатдегидрогеназы и сывороточного ферритина наряду со снижением гемоглобина, а также признаки гипокоагуляции.
2. Клиническая картина при болезни накопления эфиров холестерина (БНЭХ)
Заболевание дебютирует в возрасте старше 6 месяцев. БНЭХ характеризуется более медленным прогрессированием и, как правило, отсутствием неврологической симптоматики. ДЛКЛ является мультисистемным заболеванием, симптомы могут быть обусловлены поражением различных органов. Основными органами-мишенями являются печень, селезенка, ЖКТ, почки, сосуды. Медиана появления первых клинических симптомов заболевания составляет 5 лет, но возможна манифестация и во взрослом возрасте [13].
Заболевание проявляется гепатомегалией (больше за счет правой доли печени), связанной с накоплением ЭХ в гепатоцитах и клетках Купфера, которая определяется у 99% пациентов [2], синдромом цитолиза, дислипидемией. По мере прогрессирования заболевания формируется фиброз и цирроз печени с развитием печеночной недостаточности.
Увеличение селезенки обусловлено отложением ЭХ в макрофагах и прогрессированием цирроза печени с формированием портальной гипертензии, что сопровождается нарастанием признаков гиперспленизма (анемия, лейкопения, тромбоцитопения). Спленомегалия определяется у 74% пациентов [2, 13].
Дислипидемия проявляется повышением в сыворотке крови уровня общего холестерина, холестерина липопротеинов низкой плотности и триглицеридов при нормальном или низком уровне холестерина липопротеинов высокой плотности (гиперлипопротеинемия IIb типа), что может приводить к ускоренному развитию атеросклероза [1-4]. Гиперхолестеринемия определяется у 81% пациентов с БНЭХ, увеличение концентрации холестерина ЛПНП — у всех пациентов. Даже при терапии гиполипидемическими средствами гиперхолестеринемия сохраняется у четверти, а высокий уровень ЛПНП — у половины больных. При анализе аполипопротеинограммы у большинства пациентов выявляют повышение уровня основного аполипопротеина ЛПНП — апоВ [2].
Синдром цитолиза — увеличение уровня АСТ и АЛТ — наблюдается практически у всех пациентов и часто служит одним из первых проявлений заболевания. Активность этих ферментов в крови варьирует в широком диапазоне (от 2–5 норм до превышения данных показателей в 10–20 раз) [2].
Из-за вовлечения в патологический процесс кишечника у части больных может наблюдаться синдром мальабсорбции (недостаточность питания, диарея, стеаторея, нарушение всасывания жирорастворимых нутриентов).
При прогрессировании болезни развивается фиброз и цирроз печени, проявляющийся гепатоспленомегалией, желтухой, асцитом, варикозным расширением вен пищевода [2, 10]. Имеются сведения о 2 случаях развития гепатоцеллюлярной карциномы на фоне БНЭХ [14].
Кальцификация надпочечников, описана лишь у 5% пациентов с БНЭХ [2, 13]. Вследствие раннего развития атеросклероза часто наблюдаются ИБС, аневризма аорты и ОНМК в молодом возрасте.
При наличии у пациента признаков стеатоза печени, отрицательном алкогольном анамнезе и отсутствии стигм хронической алкогольной интоксикации (увеличение околоушных желез, контрактура Дюпюитрена и др.) необходимо проводить дифференциальную диагностику НАЖБП и ДЛКЛ (БНЭХ). Сочетание синдрома цитолиза, стеатоза печени, дислипидемии типа IIb, спленомегалии у детей и молодых лиц с нормальной массой тела может указывать на наличие БНЭХ.
Таким образом, БНЭХ следует заподозрить у молодых пациентов с гепатомегалией, увеличением АЛТ, АСТ в сыворотке крови, у которых также может иметь место повышение уровня общего холестерина и холестерина липопротеидов низкой плотности вместе с небольшим или умеренным снижением уровня холестерина липопротеидов высокой плотности и гипертриглицеридемией. (Приложение А3) [2, 13].
3. Заболевания со сходной с ДЛКЛ клинической картиной
Дифференциальная диагностика ДЛКЛ проводится с другими лизосомными болезнями накопления — болезнью Ниманна-Пика тип А, В, С, болезнью Гоше, ганглиозидозами, а также нарушениями обмена гликогена, жирных кислот, желчных кислот, болезнью Вильсона-Коновалова, семейной гиперхолестеринемией, неалкогольной жировой болезнью печени [1, 13]. Пациенты с ДЛКЛ могут длительно наблюдаться с такими диагнозами, как криптогенный цирроз печени, гепатит неясной этиологии. [1, 2,13]
Диагностика
Диагностика заболевания или состояния (группы заболеваний или состояний) медицинские показания и противопоказания к применению методов диагностики
Обращаем внимание, что, согласно требованиям к разработке клинических рекомендаций, к каждому тезису-рекомендации необходимо указывать силу рекомендаций и доказательную базу в соответствии со шкалами оценки уровня достоверности доказательств (УДД) и уровня убедительности рекомендаций (УУР). Для многих тезисов УУР и УДД будет низким по причине отсутствия посвященных им клинических исследований высокого дизайна. Невзирая на это, они являются необходимыми элементами обследования пациента для установления диагноза и выбора тактики лечения.
Критерии установления диагноза и состояния.
Определение активности ЛКЛ в сухих пятнах крови является «золотым стандартом» диагностики этого заболевания. Выявление сниженной активности ЛКЛ позволяет подтвердить диагноз.
Диагноз ДЛКЛ устанавливается на основании совокупности анамнестических и клинических данных, результатов лабораторных, инструментальных, морфологических методов исследований.
Жалобы и анамнез
При сборе анамнеза и жалоб необходимо обратить внимание на следующие жалобы и анамнестические события:
- отягощенный семейный анамнез (сходные симптомы у родных братьев и сестер пробанда, близкородственный брак);
- случаи внезапной детской смерти в семье;
- задержка физического развития;
- слабость, повышенная утомляемость;
- гепатомегалия или гепатоспленомегалия;
- диарея, боли в животе, стеаторея (при вовлеченности в патологический процесс кишечника);
- носовые кровотечения;
- желтушность кожных покровов;
- асцит;
- варикозное расширение вен пищевода (при развитии цирроза печени).
Жалобы и анамнез также описаны в разделе «клиническая картина».
Физикальное обследование
1. Физикальное обследование при инфантильной форме ДЛКЛ (болезни Вольмана)
При осмотре необходимо обратить внимание на следующие основные клинические проявления:
- увеличение размеров печени;
- увеличение размеров селезенки;
- увеличение размеров живота;
- синдром мальабсорбции;
- гипотрофия, вплоть до грубой задержки физического развития;
- прогрессирующая задержка психомоторного развития, гиперрефлексия;
- желтушность кожных покровов;
- признаки печеночной недостаточности.
2. Физикальное обследование при болезни накопления эфиров холестерина (БНЭХ), с дебютом в возрасте старше 6 месяцев
При осмотре необходимо обратить внимание на следующие клинические проявления:
- увеличение размеров печени;
- увеличение размеров селезенки.
Подробно данные физикального обследования описаны в разделе «клиническая картина».
Лабораторные диагностические исследования
1. Лабораторные диагностические исследования при инфантильной форме ДЛКЛ (болезни Вольмана)
-
Рекомендуется всем пациентам с подозрением на инфантильную форму ДЛКЛ (болезнь Вольмана) для верификации диагноза определение активности лизосомной кислой липазы в пятнах высушенной крови или лейкоцитах периферической крови. [17,18].
Уровень убедительности рекомендации В (уровень достоверности доказательств – 3).
Комментарий: Определение активности ЛКЛ в сухих пятнах крови позволяет подтвердить или опровергнуть диагноз ДЛКЛ и является «золотым стандартом» диагностики этого заболевания. Правила забора крови на карточку-фильтр приведены в Приложении А3.
-
Рекомендуется пациентам, у которых выявлено резкое снижение активности ЛКЛ в крови проведение молекулярно-генетического исследования (выявление мутаций в гене LIPA) с целью подтверждения диагноза на молекулярно-генетическом уровне, возможности проведения пренатальной и преимплантационной диагностики в семье [3,15].
Уровень убедительности рекомендации С (уровень достоверности доказательств– 5).
Комментарии: Большинство пациентов с ДЛКЛ являются гомозиготами или компаунд-гетерозиготами по мутациям в кодирующей области гена LIPA. Вместе с тем, описаны интронные мутации и протяженные перестройки гена, не выявляемые при проведении стандартного генетического исследования. Патогенность выявленных редких и новых мутаций требует дополнительных доказательств. Поскольку в настоящее время гено-фенотипическая корреляция установлена только на уровне клинической формы ДЛКЛ (БВ, либо БНЭХ), но не прослеживается для отдельных фенотипических признаков, молекулярно-генетическое исследование применяется для подтверждения, а не установления диагноза ДЛКЛ. Выявление семейной мутации гена LIPA делает возможным обследование родственников пробанда, а также проведение пренатальной и преимплантационной диагностики.
-
Рекомендуется всем пациентам с подозрением на инфантильную форму ДЛКЛ (болезнь Вольмана) проведение общего (клинического) анализа крови развернутого для выявления анемии, лейкопении, тромбоцитопении [9,13,15].
Уровень убедительности рекомендации С (уровень достоверности доказательств– 5).
Комментарии: В рамках общего (клинического) анализа крови развернутого необходимо: исследование уровня общего гемоглобина, эритроцитов, лейкоцитов, тромбоцитов в крови, дифференцированный подсчет лейкоцитов (лейкоцитарная формула), просмотр мазка крови для анализа аномалий морфологии эритроцитов, тромбоцитов и лейкоцитов, определение цветового показателя, определение размеров эритроцитов, исследование скорости оседания эритроцитов. Патогенетические механизмы формирования анемии и тромбоцитопении не до конца изучены. Одним из возможных механизмов является накопление эфиров холестерина и триглицеридов в клетках макрофагально-моноцитарной системы. Кроме того, анемия может носить алиментарный дефицитный характер, так и являться следствием гиперспленизма при формировании портальной гипертензии. Следствием гиперспленизма является также тромбоцитопения. Описаны случаи вторичного гемофагоцитарного лимфогистиоцитоза.
Учитывая крайнюю редкость БВ, ранний старт, позднюю диагностику и обычно тяжелое течение достоверных данных о необходимой частоте проведения исследования нет. Частота исследования общего (клинического) анализа крови развернутого диктуется состоянием пациента.
-
Рекомендовано проведение анализа крови биохимического общетерапевтического (исследование уровня холестерина, холестерина липопротеинов высокой плотности, холестерина липопротеинов низкой плотности, триглицеридов, общего белка, альбумина, общего билирубина, билирубина связанного (конъюгированного), билирубина свободного (неконъюгированного), определение активности аспартатаминотрансферазы, аланинаминотрансферазы, гамма-глютамилтрансферазы) при диагностике и в процессе диспансерного наблюдения всем пациентам с БВ для определения функционального состояния печени, которая является одним из органов-мишеней при БВ, и выявления дислипидемии в крови [15,16].
Уровень убедительности рекомендации С (уровень достоверности доказательств– 5)
Комментарии: для пациентов с БВ характерно значительное повышение активности трансаминаз (АЛТ, АСТ), гиперхолестеринемия, гипербилирубинемия, возможна гипертриглицеридемия; увеличение концентрации ЛПНП, у многих пациентов снижено количество ЛПВП в крови. При анализе аполипопротеинограммы у большинства пациентов выявляют повышение уровня основного аполипопротеина ЛПНП - аполипопротеина В.
Учитывая крайнюю редкость встречаемости БВ, ранний дебют, позднюю диагностику и тяжелое течение болезни достоверных данных о необходимой частоте проведения исследования нет. Частота исследования анализа крови биохимического общетерапевтического диктуется состоянием пациента. При диагностике проводится определение активности аланинаминотрансферазы, аспартатаминотрансферазы, гамма-глютамилтрансферазы в крови, исследование уровня общего билирубина, билирубина связанного (конъюгированного), холестерина, холестерина липопротеинов низкой плотности, холестерина липопротеинов высокой плотности, триглицеридов в крови. В динамике проводят определение активности аланинаминотрансферазы, аспартатаминотрансферазы, гамма-глютамилтрансферазы в крови, исследование уровня общего билирубина, билирубина связанного (конъюгированного), холестерина, холестерина липопротеинов низкой плотности, холестерина липопротеинов высокой плотности, триглицеридов в крови (не менее 1 раза в месяц).
-
Рекомендуется всем пациентам с БВ при диагностике и далее в динамике исследование коагулограммы (ориентировочного исследования системы гемостаза), определение протромбинового (тромбопластинового) времени в крови или в плазме, международного нормализованного отношения (MHO), активированного частичного тромбопластинового времени (АЧТВ) с целью своевременного выявления нарушений гемостаза. [10, 37, 45].
Уровень убедительности рекомендации С (уровень достоверности доказательств – 5)
Комментарий: при необходимости так же определяют тромбиновое время в крови, исследование уровня фибриногена в крови. Данные показатели позволяют контролировать синтетическую функцию печени, выявлять синдром печеночно-клеточной недостаточности и своевременно предупреждать осложнения, связанные с нарушением гемостаза [49].
-
Рекомендовано пациентам с БВ проводить исследование уровня общего кортизола, адренокортикотропного гормона в крови с целью своевременной диагностики надпочечниковой недостаточности и назначения заместительной терапии [15, 35].
Уровень убедительности рекомендации С (уровень достоверности доказательств – 5)
Комментарии: при выявлении признаков надпочечниковой недостаточности показано исследование уровня общего кортизола, адренокортикотропного гормона, глюкозы в крови, определение рениновой активности плазмы крови, исследование уровня натрия, калия в крови.
2. Лабораторные диагностические исследования при болезни накопления эфиров холестерина (БНЭХ)
-
Рекомендуется всем пациентам с подозрением на БНЭХ для верификации диагноза определение активности лизосомной кислой липазы в пятнах высушенной крови или лейкоцитах периферической крови [17, 18].
Уровень убедительности рекомендации С (уровень достоверности доказательств– 3).
Комментарий: Определение активности ЛКЛ в сухих пятнах крови позволяет подтвердить или опровергнуть диагноз ДЛКЛ и является «золотым стандартом» диагностики этого заболевания. Правила забора крови на карточку-фильтр приведены в Приложении А3.
-
Рекомендуется пациентам, у которых выявлено снижение активности ЛКЛ, проведение молекулярно-генетического исследования (выявление мутаций гене LIPA) с целью подтверждения диагноза на молекулярно-генетическом уровне, возможности проведения пренатальной и преимплантационной диагностики в семье [3,15].
Уровень убедительности рекомендации С (уровень достоверности доказательств– 5).
Комментарии: Большинство пациентов с ДЛКЛ являются гомозиготами или компаунд-гетерозиготами по мутациям в кодирующей области гена LIPA, описаны интронные мутации и протяженные перестройки гена, не выявляемые при проведении стандартного генетического исследования. Также патогенность выявленных редких и новых мутаций требует дополнительных доказательств. Поскольку в настоящее время гено-фенотипическая корреляция установлена только на уровне клинической формы ДЛКЛ (БВ, либо БНЭХ), но не прослеживается для отдельных фенотипических признаков, молекулярно-генетическое исследование применяется для подтверждения, а не установления диагноза ДЛКЛ. Выявление семейной мутации гена LIPA делает возможным обследование родственников пробанда, а также проведение пренатальной и преимплантационной диагностики.
-
Рекомендуется проведение общего (клинического) анализа крови, развернутого всем пациентам с подозрением на БНЭХ и далее в динамике, в среднем, не реже 1 раза в 6 месяцев с целью выявления анемии, лейкопении, тромбоцитопении [9,13,15].
Уровень убедительности рекомендации С (уровень достоверности доказательств– 5).
Комментарии: В рамках общего (клинического) анализа крови развернутого необходимо: исследование уровня общего гемоглобина, эритроцитов, лейкоцитов, тромбоцитов в крови, дифференцированный подсчет лейкоцитов (лейкоцитарная формула), просмотр мазка крови для анализа аномалий морфологии эритроцитов, тромбоцитов и лейкоцитов, определение цветового показателя, определение размеров эритроцитов, исследование скорости оседания эритроцитов. Патогенетические механизмы формирования анемии и тромбоцитопении не до конца изучены. Одним из возможных механизмов является накопление эфиров холестерина и триглицеридов в клетках макрофагально-моноцитарной системы. Кроме того, анемия может иметь как алиментарный дефицитный характер, так и являться следствием гиперспленизма. Тромбоцитопения является следствием гиперспленизма при формировании портальной гипертензии, наряду с этим описаны случаи вторичного гемофагоцитарного лимфогистиоцитоза.
-
Рекомендуется проведение анализа крови биохимического общетерапевтического (исследование уровня холестерина, холестерина липопротеинов высокой плотности, холестерина липопротеинов низкой плотности, триглицеридов, общего белка, альбумина, общего билирубина, билирубина связанного (конъюгированного), билирубина свободного (неконъюгированного), определение активности аспартатаминотрансферазы, аланинаминотрансферазы, гамма-глютамилтрансферазы) при диагностике и в процессе диспансерного наблюдения, в среднем, каждые 3-6 месяцев всем пациентам с БНЭХ для определения функционального состояния печени, которая является одним из органов-мишеней при ДЛКЛ, и определения дислипидемии в крови [15, 16, 38].
Уровень убедительности рекомендации С (уровень достоверности доказательств– 5)
Комментарии: у большинства пациентов с БНЭХ выявляют повышение активности трансаминаз (АЛТ, АСТ), гиперхолестеринемию, гипертриглицеридемию; увеличение концентрации ЛПНП, у многих пациентов снижено количество ЛПВП в крови. При анализе аполипопротеинограммы выявляют повышение уровня основного аполипопротеина ЛПНП - аполипопротеина В.
-
Рекомендуется исследование уровня мочевины, креатинина в сыворотке крови с определением скорости клубочковой фильтрации всем пациентам с БНЭХ при диагностике заболевания и при динамическом наблюдении, в среднем каждые 6-12 месяцев с целью контроля функции почек и своевременной диагностики ее нарушения [38].
Уровень убедительности рекомендации С (уровень достоверности доказательств– 5)
-
Рекомендуется всем пациентам с БНЭХ при диагностике и далее в динамике, ежегодно, исследование коагулограммы (ориентировочного исследования системы гемостаза), определение протромбинового (тромбопластинового) времени в крови или в плазме, международного нормализованного отношения (MHO), активированного частичного тромбопластинового времени (АЧТВ) с целью своевременного выявления нарушений гемостаза [10, 38, 49].
Уровень убедительности рекомендации С (уровень достоверности доказательств– 5)
Комментарии: при необходимости так же определяют тромбиновое время в крови, исследование уровня фибриногена в крови. Данные показатели позволяют контролировать синтетическую функцию печени, выявлять синдром печеночно-клеточной недостаточности и своевременно предупреждать осложнения, связанные с нарушением гемостаза [49].
Инструментальные диагностические исследования
1. Инструментальные диагностические исследования при инфантильной форме ДЛКЛ (болезни Вольмана)
-
Рекомендуется всем пациентам с подозрением на БВ и после лабораторного подтверждения диагноза проведение ультразвукового исследования (УЗИ) брюшной полости (ультразвуковое исследование органов брюшной полости (комплексное)) и забрюшинного пространства (в т.ч., ультразвуковое исследование надпочечников), а также дуплексное сканирование сосудов печени (дуплексное сканирование сосудов гепатобилиарной зоны), дуплексное сканирование сосудов селезенки с целью контроля состояния внутренних органов [2, 9, 13, 15].
Уровень убедительности рекомендации С (уровень достоверности доказательств– 5).
Комментарии: УЗИ брюшной полости позволяют определить размеры печени и селезенки, определить диаметр воротной и селезеночной вен, наличие стеатоза, фиброза или цирроза печени, кальцификатов надпочечников.
Частота проведения ультразвукового исследования органов брюшной полости (комплексного) и забрюшинного пространства (в т.ч., ультразвукового исследование надпочечников) диктуется состоянием пациента, но частота не реже 1 раза в месяц).
-
Рекомендуется пациентам с БВ при наличии признаков портальной гипертензии, проведение эзофагогастродуоденоскопии (ЭГДС) с целью оценки состояния вен пищевода и желудка [15].
Уровень убедительности рекомендации С (уровень достоверности доказательств– 5).
Комментарии: проведение ЭГДС у пациентов с фиброзом печени, признаками портальной гипертензии необходимо для оценки степени варикозного расширения вен пищевода и угрозы кровотечения.
-
Не рекомендована биопсия печени в качестве рутинного метода для подтверждения диагноза БВ [15, 19].
Уровень убедительности рекомендации С (уровень достоверности доказательств– 4).
Комментарии: может проводиться с целью оценки состояния ткани печени, определения наличия и выраженности стеатоза, стадии фиброза.
Макроскопически печень имеет яркий желто-оранжевый цвет; гистологическая картина характеризуется наличием микровезикулярного стеатоза, «пенистых» клетки Купфера, признаков фиброза и цирроза печени [13, 19].
Предпочтительно использование неинвазивных методик, например, эластографии печени (Эластометрии печени), магнитно-резонансной томографии органов брюшной полости [39].
2. Инструментальные диагностические исследования при болезни накопления эфиров холестерина (БНЭХ), с дебютом в возрасте старше 6 месяцев
-
Рекомендуется всем пациентам с подозрением на БНЭХ проведение ультразвукового исследования (УЗИ) брюшной полости (ультразвуковое исследование органов брюшной полости (комплексное)) и забрюшинного пространства, а также дуплексное сканирование сосудов печени (дуплексное сканирование сосудов гепатобилиарной зоны), дуплексное сканирование сосудов селезенки с целью динамического контроля состояния внутренних органов [2, 9, 13, 15].
Уровень убедительности рекомендации С (уровень достоверности доказательств– 5).
Комментарии: УЗИ печени и селезенки позволяют выявить увеличение печени и реже селезенки, наличие стеатоза, фиброза или цирроза печени. УЗИ забрюшинного пространства, в т.ч., УЗИ надпочечников может выявить кальцификаты надпочечников (у пациентов с БНЭХ – крайне редко, не более 5%).
-
Рекомендуется пациентам с клиническими признаками БНЭХ проведение магнитно-резонансной томографии (МРТ) органов брюшной полости и эластографии печени (эластометрии печени) с целью контроля состояния печени при проведении ФЗТ [21 – 26, 38].
Уровень убедительности рекомендации С (уровень достоверности доказательств– 5).
Комментарии: проведение оценки степени выраженности жировой дистрофии печени (по МРТ) необходимо для последующего контроля эффективности ФЗТ. Частота исследований в среднем: МРТ органов брюшной полости – 1р/год, эластографии печени – 1р/6 мес.
-
Рекомендуется пациентам с БНЭХ проведение эзофагогастродуоденоскопии (ЭГДС) с целью оценки состояния вен пищевода и желудка [15].
Уровень убедительности рекомендации С (уровень достоверности доказательств– 5).
Комментарии: проведение ЭГДС у пациентов с фиброзом печени, признаками портальной гипертензии необходимо для оценки степени варикозного расширения вен пищевода и угрозы кровотечения.
-
Рекомендуется пациентам с БНЭХ регистрация электрокардиограммы (ЭКГ), проведение эхокардиографии (ЭХОКГ) для оценки состояния сердечно-сосудистой системы, в среднем, каждые 1-2 года [2, 38].
Уровень убедительности рекомендации С (уровень достоверности доказательств– 5).
Комментарий: При наличии аритмии необходимо проведение суточного мониторирования ЭКГ (Холтеровское мониторирование), по показаниям – стресс-теста (электрокардиография с физической нагрузкой) в соответствии с рекомендациями по ведению пациентов с атеросклерозом и аритмией.
-
Рекомендуется всем пациентам с установленным диагнозом БНЭХ проведение ультразвукового допплерографического анализа сосудов головы и шеи: УЗИ брахиоцефальных структур (брахиоцефальный ствол, общая, внутренняя и наружная сонная артерии, позвоночная и подключичные артерии: оценка их диаметра, стенок, скорости кровотока, наличия/отсутствия их стенозирования, толщины комплекса интим-медиа (КИМ) для диагностики атеросклероза и оценки степени его выраженности [13, 15, 20, 38, 40, 41].
Уровень убедительности рекомендации С (уровень достоверности доказательств– 5)
Комментарий: согласно приказу от 13.10.2017 № 804н «Об утверждении номенклатуры медицинских услуг»: Дуплексное сканирование брахиоцефальных артерий с цветным допплеровским картированием кровотока, Дуплексное сканирование экстракраниальных отделов брахиоцефальных артерий, Дуплексное сканирование брахиоцефальных артерий, лучевых артерий с проведением ротационных проб.
Дополнительно может быть проведено определение лодыжечно-плечевого (лодыжечно-брахиального) индекса, исследована кальцификация коронарных артерии [38].
-
Рекомендуется взрослым пациентам с БНЭХ проведение при необходимости, по заключению врача-кардиолога, в соответствии с общими принципами подхода к диагностике атеросклероза, компьютерно-томографической ангиографии сосудов головы (компьютерно-томографическая ангиография сосудов головного мозга) и шеи (компьютерно-томографическая ангиография брахиоцефальных артерий) для оценки состояния сердечно-сосудистой системы [38].
Уровень убедительности рекомендации С (уровень достоверности доказательств–5)
-
Не рекомендована биопсия печени в качестве рутинного метода для подтверждения диагноза БНЭХ [15, 19].
Уровень убедительности рекомендации С (уровень достоверности доказательств– 5).
Комментарии: может проводиться с целью оценки состояния ткани печени, определения наличия и выраженности стеатоза, стадии фиброза.
Макроскопически печень имеет желто-оранжевый цвет; гистологическая картина характеризуется наличием микровезикулярного стеатоза, «пенистых» клетки Купфера, признаков фиброза и цирроза печени [13, 19].
Предпочтительно использование неинвазивных методик, например, УЗИ, эластографии печени (эластометрии печени), магнитно-резонансной томографии органов брюшной полости [39].
Иные диагностические исследования
1. Иные диагностические исследования при инфантильной форме ДЛКЛ (болезни Вольмана)
-
Рекомендуется в ведении пациентов с БВ на всех этапах использовать мультидисциплинарный подход в виду того, что заболевание характеризуется поражением многих органов и систем, требует комплексной терапии, что диктует необходимость совместного ведения пациента специалистами разных профилей [39, 42, 15].
Уровень убедительности рекомендации С (уровень достоверности доказательств– 5)
Комментарий: пациентов с БВ наблюдает врач-гастроэнтеролог и/или врач-педиатр при диагностике и далее не реже 1 раза в месяц, а также врач-генетик. Необходима консультация врача-диетолога с целью назначения низкожировой диеты, коррекции мальабсорбции (прекращение грудного вскармливания, подбор специализированной диеты с ограничением животных жиров, применение детской смеси с формулой триглицеридов со средней длиной цепи подбор специализированной диеты с ограничением животных жиров, а в ряде ситуаций и парентерального питания.) [2,15], консультация врача-кардиолога детского с целью оценки нарушений сердечно-сосудистой системы и терапии [2,13,15,20,26], врача - невролога с целью оценки нарушений нервной системы [14], консультация врача-трансплантолога при наличии показаний для проведения трансплантации печени. В жизнеугрожающих состояниях необходимо участие врача-анестезиолога-реаниматолога. Первичные и повторные консультации врачей иных специальностей, а также медицинского психолога показаны пациентам с БВ, имеющим нарушения функций других органов и систем.
-
Рекомендуется пациентам с установленным диагнозом БВ и наличием изменений в надпочечниках консультация врача-эндокринолога с целью оценки функции надпочечников и своевременного назначения гормонозаместительной терапии [15].
Уровень убедительности рекомендации С (уровень достоверности доказательств– 5)
2. Иные диагностические исследования при болезни накопления эфиров холестерина (БНЭХ)
-
Рекомендуется при ведении пациентов с БНЭХ на всех этапах использовать мультидисциплинарный подход в виду того, что заболевание характеризуется поражением многих органов и систем, требует комплексной терапии, что диктует необходимость совместного ведения пациента специалистами разных профилей [15, 38, 39, 42].
Уровень убедительности рекомендации С (уровень достоверности доказательств– 5)
Комментарий: пациентов с БНЭХ наблюдает врач-гастроэнтеролог, врач-терапевт (в детской практике врач-педиатр), а также врач-генетик, необходима консультация врача-диетолога с целью назначения низкожировой диеты (подбор специализированной диеты с ограничением животных жиров)[2, 15], консультация врача-кардиолога (врача-кардиолога детского) с целью оценки нарушений сердечно-сосудистой системы и терапии (оценка сосудистого риска, назначение лечения, первично и в последующем каждые 6 месяцев) [2, 13, 15, 20, 26], консультация врача-трансплантолога при наличии показаний для трансплантации печени, консультация врача-невролога при наличии показаний. В жизнеугрожающих состояниях необходимо участие врача-анестезиолога-реаниматолога.
-
Рекомендуется консультации других специалистов пациентам с подозрением на БНЭХ или с установленным диагнозом по показаниям [15].
Уровень убедительности рекомендации С (уровень достоверности доказательств– 5)
Лечение
Лечение, включая медикаментозную и немедикаментозную терапии, диетотерапию, обезболивание, медицинские показания и противопоказания к применению методов лечения
Лечение ДЛКЛ включает как патогенетическую терапию - назначение ФЗТ, так и проведение симптоматической терапии. Ведение пациентов с ДЛКЛ предполагает мультидисциплинарный подход с обязательным участием врачей-гастроэнтерологов, врачей-педиатров/врачей-терапевтов, врачей-генетиков, врачей-диетологов, врачей-кардиологов и врачей других специальностей, имеющих опыт в лечении этого редкого заболевания.
1. Патогенетическое лечение
1.1 Патогенетическое лечение при инфантильной форме ДЛКЛ (болезни Вольмана)
-
Рекомендовано проведение ферментной заместительной терапии (ФЗТ) всем пациентам с установленным диагнозом БВ с целью замедления прогрессирования заболевания, уменьшения размеров печени и селезенки, регресса или стабилизации фиброза печени, устранения дислипидемии, уменьшения накопления эфиров холестерина в органах и тканях [21-25].
Уровень убедительности рекомендации В (уровень достоверности доказательств– 2).
Комментарии: Препаратом для ФЗТ является Себелипаза альфа**. В 1 мл раствора содержится себелипазы альфа** 2 мг. Это рекомбинантная форма человеческой лизосомной кислой липазы, производимая с использованием технологии рекомбинантной ДНК в клеточной культуре куриного яйца. ФЗТ предназначена для восстановления уровня энзиматической активности, достаточного для гидролиза накопленных эфиров холестерина и для предотвращения их дальнейшего накопления. После введения себелипаза альфа** быстро выводится из системного кровотока и поглощается клетками, поступая в их лизосомы через маннозо-6-фосфатные рецепторы.
Введение препарата осуществляется парентерально в виде внутривенных инфузий. Введение осуществляется через периферический венозный катетер (катетер периферический) или через порт-систему (порт инфузионный/инъекционный, имплантируемый***), которые устанавливаются согласно методическим руководствам «Венозный доступ, 2019 (https://msestra.ru/download/file.php?id=4763) с использованием необходимых лекарственных средств. Порт-системы устанавливаются согласно Распоряжению Правительства Российской Федерации от 31.12.2018 № 3053-р «Об утверждении перечня медицинских изделий, имплантируемых в организм человека при оказании медицинской помощи в рамках программы государственных гарантий бесплатного оказания гражданам медицинской помощи, а также перечня медицинских изделий, отпускаемых по рецептам на медицинские изделия при предоставлении набора социальных услуг».
Рекомендованный режим дозирования зависит от возраста. Детям до 6 месяцев рекомендуемая начальная доза составляет 1мг/кг, в последующем, в случае недостаточного эффекта после проведения, как минимум, 4 инфузий, следует рассмотреть увеличение дозы до 3 мг/кг внутривенно 1 раз в неделю [Инструкция по медицинскому применению препарата себелипаза альфа**]. Дальнейшее увеличение дозы до 5 мг/кг 1 раз в неделю следует рассмотреть в случае недостаточного ответа после проведения, как минимум, 4 дополнительных инфузий. Последующие коррекции дозы можно проводить индивидуально в зависимости от достижения и поддержания терапевтических целей, возможно применение до 7,5 мг/кг (более высокие дозы – не изучались)[21, 43, https://grls.rosminzdrav.ru/Grls_View_v2.aspx?routingGuid=1eaa9c5e-20c6-4a75-a48c-44f5271fcf4d].
Необходимый объем препарата медленно разводят в нужном объеме раствора натрия хлорида** согласно весу (приложение А3). Весь необходимый объем раствора должен быть введен не менее чем в течение 2 часов.
Следует обращать внимание на соблюдение интервалов между инфузиями и недопустимость перерывов в терапии, т.к. нарушение режима лечения сопровождается потенциальным риском ухудшения состояния пациента и прогрессирования симптомов БВ.
Необходим мониторинг основных параметров течения заболевания: оценка окружности средней части плеча, определение активности аланинаминотрансферазы, аспартатаминотрансферазы в крови, исследование уровня ферритина в крови, исследование уровня С-реактивного белка в сыворотке крови, коагулограмма (ориентировочное исследование системы гемостаза), стойкая или ухудшающаяся органомегалия, частые интеркуррентные инфекции и стойкое ухудшение других симптомов (например, со стороны желудочно-кишечного тракта)[https://grls.rosminzdrav.ru/Grls_View_v2.aspx?routingGuid=1eaa9c5e-20c6-4a75-a48c-44f5271fcf4d].
-
Рекомендовано пациентам с БВ проведение медикаментозной премедикации антигистаминными средствами системного действия и/или нестероидными противовоспалительными и противоревматическими препаратами/препаратами группы другие анальгетики и антипиретики согласно инструкции по применению лекарственных препаратов при появлении побочных реакций с последующей инфузией себелипазы альфа** [21-25, 47, 48].
Уровень убедительности рекомендации С (уровень достоверности доказательств – 4)
Комментарии: у пациентов с БВ, получающих ФЗТ, как и при в/в введении любого другого белкового препарата, могут развиться побочные реакции (лихорадка, озноб, рвота, крапивница, тяжелые реакций гиперчувствительности аллергического типа), происходящие во время инфузии или в течение дня после проведения инфузии. При появлении побочных реакций вовремя/после инфузии рекомендуется соответствующее лечение, при котором необходимо следовать современным рекомендациям.
1.2 Патогенетическое лечение при болезни накопления эфиров холестерина (БНЭХ), в возрасте старше 6 месяцев
-
Рекомендовано проведение ферментной заместительной терапии (ФЗТ) пациентам с установленным диагнозом БНЭХ с целью замедления прогрессирования заболевания, уменьшения размеров печени и селезенки, регресса или стабилизации фиброза печени, устранения дислипидемии, уменьшения накопления эфиров холестерина в органах и тканях [21-25].
Уровень убедительности рекомендации В (уровень достоверности доказательств– 2).
Комментарии: Препаратом для ФЗТ является себелипаза альфа**. В 1 мл раствора содержится себелипазы альфа** 2 мг. Это рекомбинантная форма человеческой лизосомной кислой липазы, производимая с использованием технологии рекомбинантной ДНК в клеточной культуре куриного яйца. ФЗТ предназначена для восстановления уровня энзиматической активности, достаточного для гидролиза накопленных эфиров холестерина и для предотвращения их дальнейшего накопления. После введения себелипаза альфа** быстро выводится из системного кровотока и поглощается клетками, поступая в их лизосомы через маннозо-6-фосфатные рецепторы.
Введение препарата осуществляется парентерально в виде внутривенных инфузий. Введение осуществляется через периферический венозный катетер (катетер периферический) или через порт-систему (порт инфузионный/инъекционный, имплантируемый***), которые устанавливаются согласно методическим руководствам «Венозный доступ, 2019 (https://msestra.ru/download/file.php?id=4763) с использованием необходимых лекарственных средств. Порт-системы устанавливаются согласно Распоряжению Правительства Российской Федерации от 31.12.2018 № 3053-р «Об утверждении перечня медицинских изделий, имплантируемых в организм человека при оказании медицинской помощи в рамках программы государственных гарантий бесплатного оказания гражданам медицинской помощи, а также перечня медицинских изделий, отпускаемых по рецептам на медицинские изделия при предоставлении набора социальных услуг».
Детям старше 6 месяцев и взрослым препарат вводят из расчета 1 мг/кг каждые 2 недели. Разрешено увеличение дозы до 3 мг/кг 1 раз в 2 недели исходя из клинического ответа. Необходимый объем препарата медленно разводят в нужном объеме раствора натрия хлорида** согласно весу (приложение А3). Весь необходимый объем раствора следует вводить приблизительно в течение 2 часов. При хорошей переносимости препарата пациентом, может быть рассмотрен переход на проведение инфузии в течении 1 часа. В случае повышения дозы длительность инфузии может быть увеличена.
Следует обращать внимание на соблюдение интервалов между инфузиями и недопустимость перерывов в терапии, т.к. нарушение режима лечения сопровождается потенциальным риском ухудшения состояния пациента и прогрессирования симптомов БНЭХ.
Необходим мониторинг основных параметров течения заболевания.
При обследовании сиблингов (братьев и сестер пробанда) могут быть выявлены дети с ДЛКЛ, не имеющие клинических проявлений. Такие пациенты нуждаются в обследовании и наблюдении, начинать их лечение необходимо при появлении первых клинических или лабораторных симптомов болезни.
-
Рекомендовано пациентам с БНЭХ проведение медикаментозной премедикации антигистаминными средствами системного действия и/или нестероидными противовоспалительными и противоревматическими препаратами/препаратами группы другие анальгетики и антипиретики согласно инструкции по применению лекарственных препаратов при появлении побочных аллергических реакций с последующей инфузией себелипазы альфа** [21-25, 47, 48]. .
Уровень убедительности рекомендации С (уровень достоверности доказательств – 4)
Комментарии: у пациентов с БНЭХ, получающих ФЗТ, как и при в/в введении любого другого белкового препарата, могут развиться побочные реакции (лихорадка, озноб, рвота, крапивница, тяжелые реакции гиперчувствительности аллергического типа), происходящие во время инфузии или в течение дня проведения инфузии. При появлении побочных реакций во время/ после инфузии рекомендуется соответствующее лечение, при котором необходимо следовать современным рекомендациям.
2. Симптоматическое лечение
Включает назначение диеты с ограниченным содержанием жиров, лечение сопутствующей патологии.
2.1 Симптоматическое лечение при инфантильной форме ДЛКЛ (болезни Вольмана)
-
Рекомендовано пациентам с БВ назначение витаминов в связи с недостаточным их усвоением из пищи, обусловленным нарушением кишечного всасывания у части пациентов с БВ с целью восполнения их дефицита [15].
Уровень убедительности рекомендации С (уровень достоверности доказательств– 5)
Комментарии: для диагностики гиповитаминоза и рассмотрения вопроса о необходимости назначения терапии необходим прием (осмотр, консультация) врача-эндокринолога и/или врача-гастроэнтеролога и/или врача-диетолога.
-
Рекомендовано пациентам с БВ и надпочечниковой недостаточностью проведение заместительной терапии препаратами группы кортикостероиды системного действия (минералокортикоиды и/или глюкокортикоиды) согласно соответствующим рекомендациям с целью коррекции надпочечниковой недостаточности [15].
Уровень убедительности рекомендации С (уровень достоверности доказательств– 5)
2.2 Симптоматическое лечение при болезни накопления эфиров холестерина (БНЭХ)
-
Рекомендован рацион питания со сниженным содержанием жира пациентам с БНЭХ с целью коррекции дислипидемии [2,15].
Уровень убедительности рекомендации С (уровень достоверности доказательств– 5)
Комментарии: В РФ приняты методические рекомендации МР 2.3.1.2432-08 Нормы физиологических потребностей в энергии и пищевых веществах для различных групп населения Российской Федерации. Допускается ограничение в рационе жиров до 30% от энергетической ценности суточного рациона. Доли насыщенных, моно- и полинасыщенных жиров должны быть равными. Максимально допустимое количество холестерина - 300 мг/сут, возможно его снижение до 200 мг/сут. Уровень белка должен соответствовать физиологической норме потребления в зависимости от возраста с увеличением доли растительных белков. Рекомендуемое соотношение растительных и животных белков соответствует 1:1. Из животных белков следует отдавать предпочтение рыбе. Необходимо ограничение легкоусвояемых углеводов и увеличение сложных углеводов и растительной клетчатки. В целом, доля углеводов должна составлять 50-60% от энергетической ценности рациона. Из них 7-10% - на долю легкоусвояемых.
-
Рекомендовано пациентам с БНЭХ назначение витаминов в связи с недостаточным их усвоением, обусловленным нарушением кишечного всасывания, с целью коррекции их дефицита [15].
Уровень убедительности рекомендации С (уровень достоверности доказательств– 5)
Комментарии: для диагностики гиповитаминоза и рассмотрения вопроса о необходимости назначения терапии необходим прием (осмотр, консультация) врача-эндокринолога и/или врача-гастроэнтеролога и/или врача-диетолога.
3. Хирургическое лечение
3.1 Хирургическое лечение при инфантильной форме ДЛКЛ (болезни Вольмана)
-
Рекомендуется пациентам с БВ на стадии декомпенсированного цирроза печени, при невозможности или неэффективности проведения патогенетической терапии рассмотреть вопрос о проведении трансплантации печени [15].
Уровень убедительности рекомендации С (уровень достоверности доказательств 5)
Комментарии: Трансплантация печени также ассоциирована с высоким риском осложнений и не оказывает влияния на системные проявления заболевания. Трансплантация печени не останавливает прогрессию БВ и поражение других органов-мишеней. Кроме того, нельзя исключить риск повреждения трансплантированной печени, так как уровень ЛКЛ по-прежнему остается низким.
-
Не рекомендовано ациентам с БВ проведение ТГСК в качестве первой линии терапии [44].
Уровень убедительности рекомендации C, уровень достоверности доказательств– 5
Комментарий: Небольшое число клинических случаев, доступных для анализа, не позволяет сделать однозначного вывода об эффективности и безопасности данной терапии. Кроме того, процедура ТГСК ассоциирована с высоким риском развития осложнений, таких, например, как отторжение трансплантата, реакция «трансплантат против хозяина» и другие постоперационные осложнения. В связи с этим ТГСК не рекомендована как рутинная терапевтическая опция для пациентов с БВ [33 - 36].
3.2 Хирургическое лечение при болезни накопления эфиров холестерина (БНЭХ), с дебютом в возрасте старше 6 месяцев
-
Рекомендуется пациентам с БНЭХ на стадии декомпенсированного цирроза печени, при невозможности или неэффективности проведения патогенетической терапии рассмотреть вопрос о проведении трансплантации печени [15, 29, 38].
Уровень убедительности рекомендации С (уровень достоверности доказательств 5)
Комментарии: Трансплантация печени также ассоциирована с высоким риском осложнений и не оказывает влияния на системные проявления заболевания. Трансплантация печени не останавливает прогрессию БНЭХ и поражение других органов-мишеней. Кроме того, нельзя исключить риск повреждения трансплантированной печени, так как уровень ЛКЛ по-прежнему остается низким. [30 – 32, 38].
Медицинская реабилитация
Медицинская реабилитация, медицинские показания и противопоказания к применению методов реабилитации
Пациентам с ДЛКЛ (БВ, БНЭХ) и членам их семей необходимы консультации психолога/медицинского психолога, поскольку заболевание носит прогрессирующий характер, нужно помочь пациенту «принять» диагноз, адаптировать его к жизни для максимальной реализации его способности к обучению и самостоятельной жизни в дальнейшем [15].
Паллиативная помощь
Паллиативная помощь
Паллиативная помощь пациентам с ДЛКЛ оказывается на основании соответствующих нормативных документов (см. Приложение А3).
Прогноз
Дополнительная информация (в том числе факторы, влияющие на исход заболевания или состояния)
Прогноз при БНЭХ зависит от возраста манифестации заболевания и выраженности клинических проявлений. Своевременная диагностика и назначение патогенетической терапии на ранних стадиях заболевания определяет благоприятный прогноз и улучшает качество жизни детей с БНЭХ, предотвращая развитие цирроза печени.
При инфантильной форме ДЛКЛ (болезни Вольмана) без проведения ФЗТ прогноз неблагоприятный (летальный исход в возрасте до 6 месяцев).
Госпитализация
Организация оказания медицинской помощи
Показания для плановой госпитализации:
- проведение диагностики и лечения, требующих круглосуточного медицинского наблюдения;
- состояние, требующее активного лечения и круглосуточного медицинского наблюдения;
- состояние, требующее проведения высокотехнологичных методов лечения;
- отсутствие возможности обеспечения ФЗТ в амбулаторных и стационар замещающих условиях;
- необходимость проведения различных видов экспертиз или обследования в медицинской организации при невозможности проведения их в амбулаторных условиях, требующих динамического наблюдения (в том числе оформление заключения федерального консилиума).
Показания для экстренной госпитализации:
- острые заболевания;
- обострения хронических болезней;
- отравления и травмы, состояния, требующие интенсивной терапии и перевода в реанимационные отделения или отделения интенсивной терапии (в том числе побочные реакции, происходящие в процессе инфузии или в течение суток проведения инфузии ФЗТ, и другие угрожающие жизни острые состояния), а также круглосуточного медицинского наблюдении и проведения специальных видов обследования и лечения.
Показания к выписке пациента из медицинской организации:
- отсутствие угрозы жизни пациента;
- отсутствие угрозы развития осложнений, требующих неотложного лечения;
- стабилизация состояния и основных клинико-лабораторных показателей патологического процесса по основному заболеванию;
- отсутствие необходимости в постоянном врачебном и круглосуточном медицинском наблюдении по основному заболеванию;
- необходимости перевода пациента в другую больницу или учреждение социального обеспечения.
Принципы организации медицинской помощи пациентам с ДЛКЛ
Постановка диагноза ДЛКЛ ставит много вопросов перед родственниками пациента и перед специалистами, работающими с такими пациентами. Многочисленные проблемы, возникающие при обнаружении и развитии заболевания, для решения которых требуется грамотная организация процесса помощи при участии мультидисциплинарной команды специалистов и соблюдение основных принципов и подходов к ее оказанию.
Данные принципы должны соблюдаться на любом этапе оказания медицинской помощи, как в момент постановки диагноза, так и на любом этапе наблюдения пациента.
1) Пациент и его представители должны получать полную информацию о заболевании, его течении, патогенезе, прогнозах, осложнениях и методах возможной терапии.
2) Диагноз ДЛКЛ подразумевает возможность оказания первичной, специализированной и паллиативной помощи на всех этапах заболевания.
3) При постановке диагноза ДЛКЛ семья должна быть направлена к специалисту, имеющему опыт работы с пациентами с этим заболеванием, обладающему современной информацией о течении заболевания и возможности участия в клинических испытаниях (новых лекарственных препаратов и/или технических устройств). Также важным является информирование семей о существующих общественных организациях, работающих с этой группой пациентов.
Учитывая быстрый характер прогрессирования БВ, все дети с данным диагнозом нуждаются в стационарном наблюдении, при необходимости - с переводом в реанимационное отделение, в котором проводится лечение тяжелой прогрессирующей печеночной недостаточности согласно соответствующим клиническим рекомендациям.
- При БВ пациентов наблюдает врач-гастроэнтеролог (и/или врач-педиатр), а также врач-генетик, необходима консультация врача-диетолога с целью назначения низкожировой диеты, а в ряде ситуаций и парентерального питания.) [2,15]. Показаны консультация врача-кардиолога (врача-кардиолога детского) с целью оценки нарушений сердечно-сосудистой системы и назначения лечения [2,13,15,20,26], консультация врача-эндокринолога с целью оценки функции надпочечников и своевременного назначения гормонозаместительной терапии, врача-невролога с целью оценки нарушений со стороны нервной системы, врача-трансплантолога при наличии показаний для трансплантации печени. В жизнеугрожающих состояниях необходимо участие врача-анестезиолога-реаниматолога.
Особенности организации медицинской помощи пациентам с БНЭХ
При БНЭХ пациентов наблюдает врач-гастроэнтеролог, врач-терапевт (и/или, в детской практике, врач-педиатр) или врач общей практики (семейный врач) в среднем, 1 раз в 6-12 мес. (в соответствии с тяжестью состояния) показано комплексное обследование амбулаторно, в условиях дневного стационара и в многопрофильных стационарах.
А также врач-генетик, необходима консультация врача-диетолога с целью назначения низкожировой диеты (подбор специализированной диеты с ограничением животных жиров) [2,15], консультация врача-кардиолога (врача-кардиолога детского) с целью оценки нарушений сердечно-сосудистой системы и терапии (оценка сосудистого риска, назначение лечения) [2,13,15,20,26], консультация врача-трансплантолога при наличии показаний для трансплантации печени. В жизнеугрожающих состояниях необходимо участие врача-анестезиолога-реаниматолога.
Другие специалисты должны привлекаться по мере возникновения специфических проблем.
Профилактика
Профилактика и диспансерное наблюдение, медицинские показания и противопоказания к применению методов профилактики
1. Профилактика
-
Рекомендуется консультация врача-генетика после установления диагноза ДЛКЛ (БВ, БНЭХ) пациенту и/или его официальным представителям, с целью разъяснений генетического риска, обсуждения возможностей пренатальной и преимплантационной диагностики [15].
Уровень убедительности рекомендации С (уровень достоверности доказательств– 5).
Комментарий: Семьям с детьми с ДЛКЛ (БВ и БНЭХ) рекомендуется медико-генетическое консультирование с целью определения генетического риска. Как и при других аутосомно-рецессивных заболеваниях, при ДЛКЛ (БВ, БНЭХ) для каждой беременности риск рождения ребенка составляет 25%. В семьях, где есть ребенок с ДЛКЛ, возможно проведение пренатальной и преимплантационной диагностики. Для этого родителей необходимо направить в специализированные диагностические лаборатории и медицинские центры.
Пренатальная диагностика ДЛКЛ
-
Рекомендуется проведение пренатальной диагностики для любой последующей беременности в семьях, отягощенных хотя бы одним случаем ДЛКЛ (БВ, БНЭХ), но в случае легких форм болезни, решение о ее проведении должно быть принято после подробного обсуждения с семьей всех рисков [15].
Уровень убедительности рекомендации С (уровень достоверности доказательств– 5).
Комментарий: пренатальная диагностика проводится молекулярно-генетическими или биохимическими методами, путем исследования ДНК, выделенной из биоптата ворсин хориона на 9-11 неделе беременности и/или клеток амниотической жидкости, плодной крови на 20-22 неделе беременности.
2. Диспансерное наблюдение
2.1 Диспансерное наблюдение у пациентов с БВ
-
Рекомендован анализ крови биохимический общетерапевтический (Исследование уровня холестерина липопротеинов высокой плотности, альбумина, общего билирубина, билирубина связанного (конъюгированного), билирубина свободного (неконъюгированного), триглицеридов, холестерина, холестерина липопротеинов низкой плотности в крови, определение активности аспартатаминотрансферазы, аланинаминотрансферазы в крови, гамма-глютамилтрансферазы в крови) пациентам с БВ в процессе динамического наблюдения для определения функционального состояния печени, которая является одним из органов-мишеней при БВ, и выявления дислипидемии в крови. [15,16].
Уровень убедительности рекомендации С (уровень достоверности доказательств– 5)
Комментарии: для пациентов с БВ характерно значительное повышение активности трансаминаз (АЛТ, АСТ), гиперхолестеринемия, гипербилирубинемия, возможна гипертриглицеридемия; увеличение концентрации ЛПНП, у многих пациентов снижено количество ЛПВП в крови. При анализе аполипопротеинограммы у большинства пациентов выявляют повышение уровня основного аполипопротеина ЛПНП - аполипопротеина В.
Учитывая крайнюю редкость встречаемости БВ, ранний дебют, позднюю диагностику и тяжелое течение болезни достоверных данных о необходимой частоте проведения исследования нет. Частота исследования анализа крови биохимического общетерапевтического диктуется состоянием пациента.
В динамике проводят определение активности аланинаминотрансферазы, аспартатаминотрансферазы в крови, исследование уровня общего билирубина, билирубина связанного (конъюгированного), холестерина, холестерина липопротеинов низкой плотности, холестерина липопротеинов высокой плотности, триглицеридов в крови (не менее 1 раза в месяц).
-
Рекомендуется всем пациентам с БВ в динамике проведение коагулограммы (ориентировочного исследования системы гемостаза), определение протромбинового (тромбопластинового) времени в крови или в плазме, международного нормализованного отношения (MHO), активированного частичного тромбопластинового времени (АЧТВ) с целью своевременного выявления нарушений гемостаза. [10, 37, 45].
Уровень убедительности рекомендации С (уровень достоверности доказательств – 4)
Комментарий: при необходимости так же определяют тромбиновое время в крови, исследование уровня фибриногена в крови. Данные показатели позволяют контролировать синтетическую функцию печени, выявлять синдром печеночно-клеточной недостаточности и своевременно предупреждать осложнения, связанные с нарушением гемостаза [49].
-
Рекомендовано проведение ультразвукового исследования (УЗИ) брюшной полости (Ультразвуковое исследование органов брюшной полости (комплексное)) и забрюшинного пространства (в т.ч., Ультразвуковое исследование надпочечников), а также Дуплексное сканирование сосудов печени (Дуплексное сканирование сосудов гепатобилиарной зоны), Дуплексное сканирование сосудов селезенки всем пациентам с БВ с целью контроля состояния внутренних органов [2, 9, 13, 15].
Уровень убедительности рекомендации С (уровень достоверности доказательств– 5).
Комментарии: УЗИ брюшной полости позволяют определить размеры печени и селезенки, определить диаметр воротной и селезеночной вен, наличие стеатоза, фиброза или цирроза печени, кальцификатов надпочечников.
Частота проведения ультразвукового исследования органов брюшной полости (комплексного) и забрюшинного пространства (в т.ч., ультразвукового исследование надпочечников) диктуется состоянием пациента, но частота не менее 1 раза в месяц)
-
Рекомендовано пациентам с БВ при наличии признаков портальной гипертензии проведение эзофагогастродуоденоскопии (ЭГДС) для оценки состояния вен пищевода и желудка [15].
Уровень убедительности рекомендации С (уровень достоверности доказательств– 5).
Комментарии: проведение ЭГДС у пациентов с фиброзом печени, признаками портальной гипертензии необходимо для оценки степени варикозного расширения вен пищевода и угрозы кровотечения.
-
Рекомендуется в ведении пациентов с БВ на всех этапах использовать мультидисциплинарный подход в виду того, что заболевание характеризуется поражением многих органов и систем, требует комплексной терапии, что диктует необходимость совместного ведения пациента специалистами разных профилей [39, 42, 15].
Уровень убедительности рекомендации С (уровень достоверности доказательств– 5)
Комментарий: пациентов с БВ наблюдает врач-гастроэнтеролог и/или врач-педиатр (не реже 1 раза в месяц), а также врач-генетик, необходима консультация врача-диетолога с целью назначения низкожировой диеты, коррекции мальабсорбции (подбор специализированной диеты с ограничением животных жиров, а в ряде ситуаций и парентерального питания.) [2,15], консультация врача-кардиолога детского с целью оценки нарушений сердечно-сосудистой системы и терапии [2,13,15,20,26], врача - невролога с целью оценки нарушений нервной системы [14], консультация врача-трансплантолога при наличии показаний для проведения трансплантации печени. В жизнеугрожающих состояниях необходимо участие врача-анестезиолога-реаниматолога.
2.2 Диспансерное наблюдение у пациентов с БНЭХ
-
Рекомендуется проведение общего (клинического) анализа крови, развернутого всем пациентам с БНЭХ в динамике, в среднем, не реже 1 раза в 6 месяцев с целью выявления анемии, лейкопении, тромбоцитопении [9,13,15].
Уровень убедительности рекомендации С (уровень достоверности доказательств– 5).
Комментарии: В рамках общего (клинического) анализа крови развернутого необходимо: исследование уровня общего гемоглобина, эритроцитов, лейкоцитов, тромбоцитов в крови, дифференцированный подсчет лейкоцитов (лейкоцитарная формула), просмотр мазка крови для анализа аномалий морфологии эритроцитов, тромбоцитов и лейкоцитов, определение цветового показателя, определение размеров эритроцитов, исследование скорости оседания эритроцитов. Патогенетические механизмы формирования анемии и тромбоцитопении не до конца изучены. Одним из возможных механизмов является накопление эфиров холестерина и триглицеридов в клетках макрофагально-моноцитарной системы. Кроме того, анемия может иметь как алиментарный дефицитный характер, так и являться следствием гиперспленизма. Тромбоцитопения является следствием гиперспленизма при формировании портальной гипертензии, наряду с этим описаны случаи вторичного гемофагоцитарного лимфогистиоцитоза.
-
Рекомендован анализ крови биохимический общетерапевтический всем пациентам с БНЭХ в процессе динамического наблюдения, в среднем, каждые 3-6 мес. для определения функционального состояния печени, которая является одним из органов-мишеней при ДЛКЛ, и определения дислипидемии в крови. Проводят определение активности аланинаминотрансферазы, аспартатаминотрансферазы, гамма-глютамилтрансферазы в крови, щелочной фосфатазы в крови, исследование уровня общего билирубина в крови, билирубина связанного (конъюгированного), холестерина в крови, холестерина липопротеинов высокой плотности, холестерина липопротеинов низкой плотности, триглицеридов в крови, альбуминов в крови. [15, 16, 38].
Уровень убедительности рекомендации С (уровень достоверности доказательств– 5)
Комментарии: у большинства пациентов с БНЭХ выявляют повышение активности трансаминаз (АЛТ, АСТ), гиперхолестеринемию, гипертриглицеридемию; увеличение концентрации ЛПНП, у многих пациентов снижено количество ЛПВП в крови. При анализе аполипопротеинограммы выявляют повышение уровня основного аполипопротеина ЛПНП - аполипопротеина В.
-
Рекомендовано всем пациентам с БНЭХ в процессе динамического наблюдения, ежегодно, проведение коагулограммы (ориентировочного исследования системы гемостаза): определение протромбинового (тромбопластинового) времени в крови или в плазме и международного нормализованного отношения (MHO), активированного частичного тромбопластинового времени (АЧТВ) с целью своевременного выявления нарушений гемостаза [10, 38, 49].
Уровень убедительности рекомендации С (уровень достоверности доказательств – 4)
Комментарий: при необходимости так же определяют тромбиновое время в крови, исследование уровня фибриногена в крови. Данные показатели позволяют контролировать синтетическую функцию печени, выявлять синдром печеночно-клеточной недостаточности и своевременно предупреждать осложнения, связанные с нарушением гемостаза [49].
-
Рекомендовано проведение ультразвукового исследования (УЗИ) брюшной полости (Ультразвуковое исследование органов брюшной полости (комплексное)) и забрюшинного пространства, а также Дуплексное сканирование сосудов печени (Дуплексное сканирование сосудов гепатобилиарной зоны), Дуплексное сканирование сосудов селезенки всем пациентам с БНЭХ с целью динамического контроля состояния внутренних органов [2, 9, 13, 15].
Уровень убедительности рекомендации С (уровень достоверности доказательств – 5)
Комментарии: УЗИ печени и селезенки позволяют выявить увеличение печени и реже селезенки, наличие стеатоза, фиброза или цирроза печени. УЗИ забрюшинного пространства, в т.ч., УЗИ надпочечников может выявить кальцификаты надпочечников (у пациентов с БНЭХ – крайне редко, не более 5%).
-
Рекомендовано пациентам с БНЭХ проведение магнитно-резонансной томографии (МРТ) органов брюшной полости и эластографии печени (эластометрии печени) с целью контроля состояния печени при проведении ФЗТ [21 – 26, 38].
Уровень убедительности рекомендации С (уровень достоверности доказательств – 5)
Комментарии: для оценки степени выраженности жировой дистрофии печени (по МРТ) необходимо для последующего контроля эффективности ФЗТ. Частота исследований в среднем: МРТ органов брюшной полости – 1р/год, эластографии печени – 1р/6 мес.
-
Рекомендовано пациентам с БНЭХ проведение эзофагогастродуоденоскопии (ЭГДС) при наличии признаков портальной гипертензии, жалоб на боли в животе или наличие сопутствующей гастроэнтерологической патологии с целью оценки состояния верхних отделов ЖКТ [15].
Уровень убедительности рекомендации С (уровень достоверности доказательств – 5)
Комментарии: проведение ЭГДС у пациентов с фиброзом печени, признаками портальной гипертензии необходимо для оценки степени варикозного расширения вен пищевода и угрозы кровотечения.
-
Рекомендовано пациентам с БНЭХ регистрация электрокардиографии (ЭКГ), проведение эхокардиографии (ЭХОКГ) для оценки состояния сердечно-сосудистой системы, в среднем, каждые 1-2 года [2, 38].
Уровень убедительности рекомендации С (уровень достоверности доказательств – 5)
Комментарий: При наличии аритмии необходимо проведение суточного мониторирования ЭКГ (Холтеровское мониторирование), по показаниям - стресс-теста (Электрокардиография с физической нагрузкой) в соответствии с рекомендациями по ведению пациентов с атеросклерозом и аритмией.
-
Рекомендуется при ведении пациентов с БНЭХ на всех этапах использовать мультидисциплинарный подход в виду того, что заболевание характеризуется поражением многих органов и систем, требует комплексной терапии, что диктует необходимость совместного ведения пациента специалистами разных профилей [15, 38, 39, 42].
Уровень убедительности рекомендации С (уровень достоверности доказательств – 5)
Комментарий: пациентов с БНЭХ наблюдает врач-гастроэнтеролог, врач-терапевт (в детской практике врач-педиатр), а также врач-генетик, необходима консультация врача-диетолога с целью назначения низкожировой диеты (подбор специализированной диеты с ограничением животных жиров) [2, 15], консультация врача-кардиолога (врача-кардиолога детского) с целью оценки нарушений сердечно-сосудистой системы и терапии (оценка сосудистого риска, назначение лечения, первично и в последующем каждые 6 месяцев) [2, 13, 15, 20, 26], консультация врача-трансплантолога при наличии показаний для трансплантации печени. При наличии неврологических нарушений: консультация врача-невролога (первично и далее ежегодно). В жизнеугрожающих состояниях необходимо участие врача-анестезиолога-реаниматолога. Первичные и повторные консультации врачей иных специальностей, а также медицинского психолога показаны пациентам с БВ, имеющим нарушения функций других органов и систем.
Информация
Источники и литература
-
Клинические рекомендации Российского общества медицинских генетиков
- Valayannopoulos V, Mengel E, Brassier A, Grabowski G. Lysosomal acid lipase deficiency: Expanding differential diagnosis. Mol Genet Metab. 2017;120(1-2):62-66. doi: 10.1016/j.ymgme.2016.11.002 Bernstein DL, Hülkova H, Bialer MG, Desnick RJ. Cholesteryl ester storage disease: review of the findings in 135 reported patients with an underdiagnosed disease. J Hepatol. 2013;58(6):1230-1243. doi: 10.1016/j.jhep.2013.02.014 Grabowski GA, Charnas L, Du H. Lysosomal acid lipase deficiencies: the Wolman disease/cholesteryl ester storage disease spectrum. In: Scriver Valle D, Beaudet AL, Vogelstein B, Kinzler KW, Antonarakis SE, Ballabio A,editors. Metabolic and molecular bases of inherited disease – OMMBID. New York: McGraw-Hill; 2014 Fouchier SW, Defesche JC. Lysosomal acid lipase A and the hypercholesterolaemic phenotype. Curr Opin Lipidol. 2013;24(4):332-338. doi:10.1097/MOL.0b013e328361f6c6 Pericleous M, Kelly C, Wang T, Livingstone C, Ala A. Wolman"s disease and cholesteryl ester storage disorder: the phenotypic spectrum of lysosomal acid lipase deficiency. Lancet Gastroenterol Hepatol. 2017;2(9):670-679. doi:10.1016/S2468-1253(17)30052-3 Santos Silva E, Klaudel-Dreszler M, Bakuła A, et al. Early onset lysosomal acid lipase deficiency presenting as secondary hemophagocytic lymphohistiocytosis: Two infants treated with sebelipase alfa. Clin Res Hepatol Gastroenterol. 2018;42(5):e77-e82. doi:10.1016/j.clinre.2018.03.012 Sadhukhan M, Saha A, Vara R, Bhaduri B. Infant case of lysosomal acid lipase deficiency: Wolman"s disease. BMJ Case Rep. 2014;2014:bcr2013202652. Published 2014 May 15. doi:10.1136/bcr-2013-202652 Каменец Е.А., Печатникова Н.Л., Какаулина В.С., Михайлова С.В., Строкова Т.В., Жаркова М.С., Потехин О.Е., Захарова Е.Ю. Дефицит лизосомной кислой липазы у российских больных: молекулярная характеристика и эпидемиология. Медицинская генетика. 2019;18(8):3-16. Дегтярева А.В., Пучкова А.А., Жданова С.И., Дегтярев Д.Н. Болезнь Вольмана - тяжелая младенческая форма дефицита лизосомной кислой липазы // Неонатология: новости, мнения, обучение. 2019. Т. 7. № 2. С. 42-51. Jones SA, Valayannopoulos V, Schneider E, et al. Rapid progression and mortality of lysosomal acid lipase deficiency presenting in infants. Genet Med. 2016;18(5):452-458. doi:10.1038/gim.2015.108 Маевская, М. В., Ивашкин, В. Т., Жаркова, М. С., Некрасова, Т. П., Аюшева, Г. И., & Масленников, Р. В. (2016). Редкие формы неалкогольной жировой болезни печени: наследственный дефицит лизосомной кислой липазы. Гепатология, 3, 41–51. Desai PK, Astrin KH, Thung SN, et al. Cholesteryl ester storage disease: pathologic changes in an affected fetus. Am J Med Genet. 1987;26(3):689-698. doi:10.1002/ajmg.1320260324 Decarlis S, Agostoni C, Ferrante F, Scarlino S, Riva E, Giovannini M. Combined hyperlipidaemia as a presenting sign of cholesteryl ester storage disease. J Inherit Metab Dis. 2009;32 Suppl 1:S11-S13. doi:10.1007/s10545-008-1027-2 Riva S, Spada M, Sciveres M, et al. Hepatocarcinoma in a child with cholesterol ester storage disease. Dig Liver Dis. 2008;40(9):784. doi: 10.1016/j.dld.2008.01.009 Hoffman EP, Barr ML, Giovanni MA, et al. Lysosomal Acid Lipase Deficiency. 2015 Jul 30 [Updated 2016 Sep 1]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2020. Available from: https://www.ncbi.nlm.nih.gov/books/NBK305870/ Quinn AG, Burton B, Deegan P et al. Sustained elevations in LDL cholesterol and serum transaminases from early childhood are common in lysosomal acid lipase deficiency. Mol Genet Metab 2014;111:S89 Hamilton J, Jones I, Srivastava R, Galloway P. A new method for the measurement of lysosomal acid lipase in dried blood spots using the inhibitor Lalistat 2. Clin Chim Acta. 2012;413(15-16):1207-1210. doi: 10.1016/j.cca.2012.03.019 Lukacs Z, Barr M, Hamilton J. Best practice in the measurement and interpretation of lysosomal acid lipase in dried blood spots using the inhibitor Lalistat 2. Clin Chim Acta. 2017; 471:201-205. doi: 10.1016/j.cca.2017.05.027 Hůlková H, Elleder M. Distinctive histopathological features that support a diagnosis of cholesterol ester storage disease in liver biopsy specimens. Histopathology. 2012;60(7):1107-1113. doi:10.1111/j.1365-2559.2011.04164.x Strebinger G, Müller E, Feldman A, Aigner E. Lysosomal acid lipase deficiency - early diagnosis is the key. Hepat Med. 2019;11:79-88. Published 2019 May 23. doi:10.2147/HMER.S201630 Jones SA, Rojas-Caro S, Quinn AG, et al. Survival in infants treated with sebelipase Alfa for lysosomal acid lipase deficiency: an open-label, multicenter, dose-escalation study. Orphanet J Rare Dis. 2017;12(1):25. Published 2017 Feb 8. doi:10.1186/s13023-017-0587-3 Burton BK, Balwani M, Feillet F, et al. A Phase 3 Trial of Sebelipase Alfa in Lysosomal Acid Lipase Deficiency. N Engl J Med. 2015;373(11):1010-1020. doi:10.1056/NEJMoa1501365 Abel F., Arnoux J.B., Kostyleva M. et al. Benefit of Sebelipase Alfa in Children and Adults With Lysosomal Acid Lipase Deficiency Based on Analysis of Efficacy Overall and by Baseline Alanine Aminotransferase Level // J. of Hepatology. Vol. 64 (2). P.298–299. Su K, Donaldson E, Sharma R. Novel treatment options for lysosomal acid lipase deficiency: critical appraisal of sebelipase alfa. Appl Clin Genet. 2016;9:157-167. Published 2016 Oct 17. doi:10.2147/TACG.S86760 Valayannopoulos V, Malinova V, Honzík T, et al. Sebelipase alfa over 52 weeks reduces serum transaminases, liver volume and improves serum lipids in patients with lysosomal acid lipase deficiency. J Hepatol. 2014;61(5):1135-1142. doi: 10.1016/j.jhep.2014.06.022 Maciejko JJ. Managing Cardiovascular Risk in Lysosomal Acid Lipase Deficiency [published correction appears in Am J Cardiovasc Drugs. 2017 Jun;17 (3):233]. Am J Cardiovasc Drugs. 2017;17(3):217-231. doi:10.1007/s40256-017-0216-5 В.Е.Новиков, Е.И.Климкина// Фармакология гепатопротекторов// Обз. клин.фармакол. лек.тер.-2005-Т.4- №1.-с.2-20. Строкова Т.В., Багаева М.Э., Матинян И.А. Дефицит лизосомной кислой липазы РМЖ . 2017. 25(19):1346-1351 Лаврова А.Е., Коновалова Е.Ю., Давыдова Д.А., Шеляхин В.Е.,Лобанова Е.В. Дефицит лизосомной кислой липазы у ребенка 5 лет Педиатрия. Журнал им. Г.Н. Сперанского. 2017. Т. 96. № 6. С. 183-186. Kale AS, Ferry GD, Hawkins EP. End-stage renal disease in a patient with cholesteryl ester storage disease following successful liver transplantation and cyclosporine immunosuppression. J Pediatr Gastroenterol Nutr. 1995;20(1):95-97. doi:10.1097/00005176-199501000-00016 Ambler GK, Hoare M, Brais R, et al. Orthotopic liver transplantation in an adult with cholesterol ester storage disease. JIMD Rep. 2013; 8:41-46. doi:10.1007/8904_2012_155 Bernstein DL, Lobritto S, Iuga A, et al. Lysosomal acid lipase deficiency allograft recurrence and liver failure- clinical outcomes of 18 liver transplantation patients. Mol Genet Metab. 2018;124(1):11-19. doi: 10.1016/j.ymgme.2018.03.010 Gramatges MM, Dvorak CC, Regula DP, Enns GM, Weinberg K, Agarwal R. Pathological evidence of Wolman"s disease following hematopoietic stem cell transplantation despite correction of lysosomal acid lipase activity. Bone Marrow Transplant. 2009;44(7):449-450. doi:10.1038/bmt.2009.57 Stein J, Garty BZ, Dror Y, Fenig E, Zeigler M, Yaniv I. Successful treatment of Wolman disease by unrelated umbilical cord blood transplantation. Eur J Pediatr. 2007;166(7):663-666. doi:10.1007/s00431-006-0298-6 Tolar J, Petryk A, Khan K, et al. Long-term metabolic, endocrine, and neuropsychological outcome of hematopoietic cell transplantation for Wolman disease. Bone Marrow Transplant. 2009;43(1):21-27. doi:10.1038/bmt.2008.273 Yanir A, Allatif MA, Weintraub M, Stepensky P. Unfavorable outcome of hematopoietic stem cell transplantation in two siblings with Wolman disease due to graft failure and hepatic complications. Mol Genet Metab. 2013;109(2):224-226. doi: 10.1016/j.ymgme.2013.03.007 Cohen JL, Burfield J, Valdez-Gonzalez K, et al. Early diagnosis of infantile-onset lysosomal acid lipase deficiency in the advent of available enzyme replacement therapy. Orphanet J Rare Dis. 2019;14(1):198. Published 2019 Aug 14. doi:10.1186/s13023-019-1129-y Kohli R, Ratziu V, Fiel MI, Waldmann E, Wilson DP, Balwani M. Initial assessment and ongoing monitoring of lysosomal acid lipase deficiency in children and adults: Consensus recommendations from an international collaborative working group. Mol Genet Metab. 2020;129(2):59-66. doi: 10.1016/j.ymgme.2019.11.004 Harrison SA. Management of Lysosomal Acid Lipase Deficiency for the Gastroenterologist and Hepatologist. Gastroenterol Hepatol (N Y). 2016;12(5):331-333. Dixon DB. Non-Invasive Techniques in Pediatric Dyslipidemia. In: Feingold KR, Anawalt B, Boyce A, et al., eds. Endotext. South Dartmouth (MA): MDText.com, Inc.; September 8, 2020. Zharkova M, Nekrasova T, Ivashkin V, Maevskaya M, Strokova T. Fatty Liver and Systemic Atherosclerosis in a Young, Lean Patient: Rule Out Lysosomal Acid Lipase Deficiency. Case Rep Gastroenterol. 2019;13(3):498-507. Published 2019 Dec 4. doi:10.1159/000504646 Erwin AL. The role of sebelipase alfa in the treatment of lysosomal acid lipase deficiency. Therap Adv Gastroenterol. 2017;10(7):553-562. doi:10.1177/1756283X17705775 Jones S.A., AlSayed M., Broomfield A.A. et al. Management guidelines for infantile onset lysosomal acid lipase deficiency (LALD). Mol. Genet. Metab. 2018;123(2):S72–S73 Pastores GM, Hughes DA. Lysosomal Acid Lipase Deficiency: Therapeutic Options. Drug Des Devel Ther. 2020;14:591-601. Published 2020 Feb 11. doi:10.2147/DDDT.S149264 Al Essa M, Nounou R, Sakati N, et al. Wolman"s disease: The King Faisal Specialist Hospital and Research Centre experience. Ann Saudi Med. 1998;18(2):120-124. doi:10.5144/0256-4947.1998.120 Witeck CDR, Schmitz AC, de Oliveira JMD, Porporatti AL, De Luca Canto G, Pires MMS. Lysosomal acid lipase deficiency in pediatric patients: a scoping review. J Pediatr (Rio J). 2022 Jan-Feb;98(1):4-14. doi: 10.1016/j.jped.2021.03.003. Malinová V, Balwani M, Sharma R, et al. Sebelipase alfa for lysosomal acid lipase deficiency: 5-year treatment experience from a phase 2 open-label extension study. Liver Int. 2020;40(9):2203-2214. doi:10.1111/liv.14603 Attachment 2 KANUMA - Sebelipase - Alexion Pharmaceuticals Australia Pty Ltd - PM-2016-01313-1-3 -Extract from the CER FINAL 14 June 2018 Клиническая лабораторная диагностика заболеваний печени и желчевыводящих путей: руководство для врачей / А.И. Карпищенко и др. под ред. А.И. Карпищенко. – М.: ГЭОТАР-Медиа, 2020. – 464 c
-
Клинические рекомендации Российского трансплантологического общества
-
Клинические рекомендации Российской гастроэнтерологической ассоциации
-
Клинические рекомендации Союза педиатров России
Информация
Список сокращений
АЛТ – аланинаминотрансфераза
АСТ – аспартатаминотрансфераза
АпоВ – аполипопротеин В
БНЭХ – болезнь накопления эфиров холестерина
ГМГ-КоА – гидроксиметилглутарил-коэнзим А
ДЛКЛ – дефицит лизосомной кислой липазы
КИМ – комплекс интима медиа
ЛКЛ – лизосомная кислая липаза
ЛПНП – липопротеины низкой плотности
ЛПВП – липопротеины высокой плотности
ЛПОНП – липопротеины очень низкой плотности
МРТ – магнитно-резонансная томография
НАЖБП – неалкогольная жировая болезнь печени
НАСГ – неалкогольный стеатогепатит
ОНМК – острое нарушение мозгового кровообращения
ПОЛ – перекисное окисление липидов
СРЭСБ – стериновый регуляторный элемент связывания белков
ТГ – триглицериды
ТГСК – трансплантация гемопоэтических стволовых клеток
УДХК – урсодезоксихолевая кислота
УЗДГ– ультразвуковая допплерография
УЗИ – ультразвуковое исследование
ФЗТ – ферментная заместительная терапия
ЭГДС – эзофагогастродуоденоскопия
ЭКГ – электрокардиограмма
ЭХ – эфиры холестерина
ЭХО-КГ– эхокардиография
Термины и определения
Лизосомные болезни накопления — группа наследственных моногенных заболеваний, связанных с нарушением функции лизосом.
Ферментная заместительная терапия — лечение, заключающееся во введении ферментов (в том числе и рекомбинантных) пациентам с наследственным дефектом метаболизма.
Критерии оценки качества медицинской помощи
при оказании специализированной медицинской помощи
Критерии качества при БВ
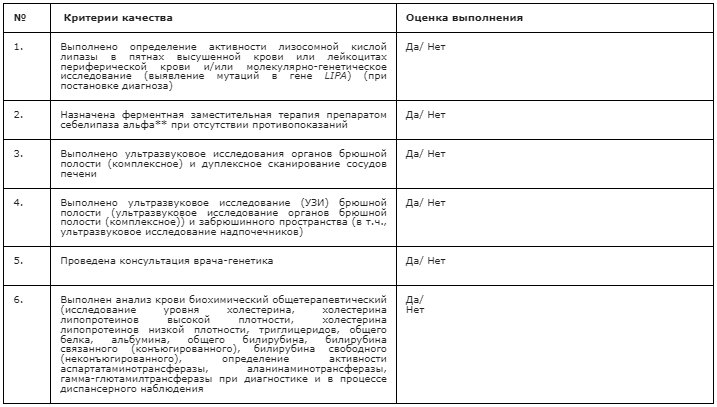
Критерии качества при БНЭХ
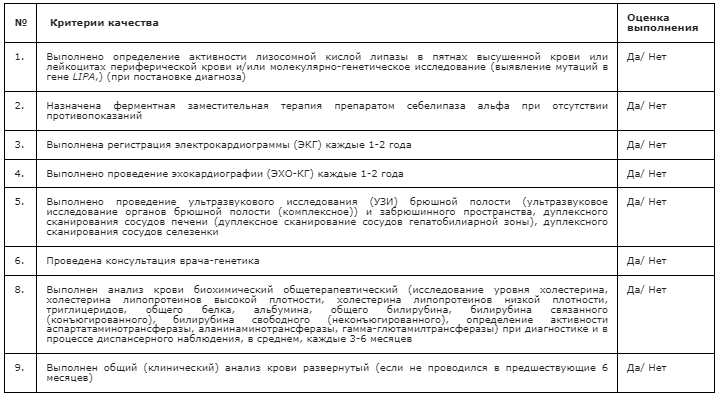
Приложение А1. Состав рабочей группы по разработке и пересмотру клинических рекомендаций
1. Анисимова Инга Вадимовна, к.м.н., заведующая отделом организации медицинской помощи-врач-генетик ФГБНУ «Медико-генетический научный центр им. академика Н.П. Бочкова», член Ассоциации медицинских генетиков.
2. Албегова Марина Бечербиевна, к.м.н., заведующая консультативно-педиатрическим отделением отдела Педиатрии ФГБУ «НМИЦ акушерства, гинекологии и перинатологии им. Ак. В.И. Кулакова»
3. Багаева Мадлена Энверовна, к.м.н., старший научный сотрудник отделения педиатрической гастроэнтерологии, гепатологии и диетотерапии Клиники ФГБУН "ФИЦ питания и биотехнологии"
4. Байдакова Галина Викторовна, к.б.н., ведущий научный сотрудник лаборатории наследственных болезней обмена ФГБНУ "Медико-генетический научный центр им. академика Н.П.Бочкова", член Российского общества медицинских генетиков.
5. Баранов Александр Александрович, академик РАН, профессор, д.м.н., Почетный Президент Союза педиатров России, советник руководителя НИИ педиатрии и охраны здоровья детей ЦКБ РАН, главный внештатный специалист педиатр Минздрава России.
6. Вашакмадзе Нато Джумберовна, д.м.н., заведующая отделом редких болезней НИИ педиатрии и охраны здоровья детей ЦКБ РАН, член Союза педиатров России
7. Вишнева Елена Александровна, д.м.н., заместитель руководителя по науке НИИ педиатрии и охраны здоровья детей ЦКБ РАН, доцент кафедры факультетской педиатрии педиатрического факультета ФГБОУ ВО «РНИМУ им. Н.И. Пирогова» Минздрава России
8. Гундобина Ольга Станиславовна, к.м.н., врач- гастроэнтеролог НИИ педиатрии и охраны здоровья детей ФГБУЗ ЦКБ РАН, член Союза педиатров России
9. Дегтярева Анна Владимировна, д.м.н., профессор, заведующая отделом педиатрии, ФГБУ «НМИЦ акушерства, гинекологии и перинатологии им. Ак. В.И. Кулакова», профессор кафедры неонатологии Института здоровья детей ФГАОУ ВО "Первый Московский государственный медицинский университет им. И.М. Сеченова" Минздрава России (Сеченовский университет).
10. Ежов Марат Владиславович, д.м.н., профессор, главный научный сотрудник отдела проблем атеросклероза НИИ клинической кардиологии им. А.Л. Мясникова ФГБУ «НМИЦК» МЗ РФ
11. Жаркова Мария Сергеевна, к. м. н., заведующая отделением гепатологии клиники пропедевтики внутренних болезней, гастроэнтерологии, гепатологии им. В.Х. Василенко, Университетская Клиническая Больница №2, ФГАОУ ВО «Первый МГМУ им. И.М. Сеченова» Минздрава России (Сеченовский университет)
12. Журкова Наталия Вячеславовна, к.м.н., ведущий научный сотрудник Отдел орфанных болезней и профилактики инвалидизирующих заболеваний, врач-генетик НИИ педиатрии и охраны здоровья детей ФГБУЗ ЦКБ РАН, член Ассоциации медицинских генетиков (АМГ).
13. Захарова Екатерина Юрьевна, д.м.н., заведующая лабораторией наследственных болезней обмена ФГБНУ "Медико-генетический научный центр им. академика Н.П.Бочкова", член Российского общества медицинских генетиков, член европейского общества по изучению наследственных болезней обмена веществ (SSIEM).
14. Ивашкин Владимир Трофимович, академик РАН, профессор, д.м.н., заведующий кафедрой пропедевтики внутренних болезней, директор Клиники пропедевтики внутренних болезней, гастроэнтерологии и гепатологии им. В.Х. Василенко ФГАОУ ВО «Первый МГМУ им. И.М. Сеченова» Минздрава России (Сеченовский университет)
15. Каменец Елена Анатольевна, сотрудник лаборатории наследственных болезней обмена ФГБНУ "Медико-генетический научный центр им. академика Н.П. Бочкова", член Российского общества медицинских генетиков.
16. Куцев Сергей Иванович, академик РАН, д.м.н., директор ФГБНУ "Медико-генетический научный центр им. академика Н.П. Бочкова ", Президент Ассоциации медицинских генетиков (АМГ).
17. Лаврова Алла Евгеньевна, д.м.н., главный научный сотрудник, врач высшей квалификационной категории по специальности “Педиатрия”, заведующий 2-м м педиатрическим отделением с медицинской реабилитацией, ФГБОУ ВО «Приволжский исследовательский медицинский университет» МЗ РФ
18. Матинян Ирина Александровна, к.м.н., научный сотрудник отделения педиатрической гастроэнтерологии, гепатологии и диетотерапии Клиники ФГБУН "ФИЦ питания и биотехнологии"
19. Михайлова Светлана Витальевна, д.м.н., заведующая отделением ФГБУ «Российская Детская Клиническая Больница» МЗ РФ.
20. Намазова-Баранова Лейла Сеймуровна, академик РАН, профессор, д.м.н., Президент Союза педиатров России; паст-президент EPA/UNEPSA; руководитель НИИ педиатрии и охраны здоровья детей ЦКБ РАН, заведующая кафедрой факультетской педиатрии педиатрического факультета ФГБОУ ВО «РНИМУ им. Н.И. Пирогова» Минздрава России, главный внештатный детский специалист по профилактической медицине Минздрава России
21. Пашкова Ирина Евгеньевна, к.м.н., заведующая педиатрическим отделением ФГБУ "НМИЦ ТИО им. ак. В.И. Шумакова" МЗ РФ
22. Петряйкина Елена Ефимовна, д.м.н., директор РДКБ ФГАОУ ВО РНИМУ им. Н.И. Пирогова Минздрава России, профессор кафедры факультетской педиатрии педиатрического факультета РНИМУ имени Н.И. Пирогова, профессор кафедры доказательной медицины медицинского института РУДН, Главный внештатный специалист детский эндокринолог Департамента здравоохранения города Москвы
23. Первунина Татьяна Михайловна, к.м.н., Директор Института перинатологии и педиатрии, ФГБУ НМИЦ им В.А. Алмазова МЗ РФ
24. Печатникова Наталья Леонидовна, врач-невролог, заведующий отделением, Морозовская ДГКБ ДЗМ
25. Репина Светлана Афанасьевна, к.м.н., врач-генетик ФГБНУ «Медико-генетический научный центр им. академика Н.П. Бочкова», член Российского общества медицинских генетиков, член Ассоциации медицинских генетиков.
26. Селимзянова Лилия Робертовна, к.м.н., ведущий научный сотрудник НИИ педиатрии и охраны здоровья детей ЦКБ РАН Министерства науки и высшего образования Российской Федерации, доцент кафедры педиатрии и детской ревматологии ФГАОУ «Первый МГМУ им. И.М. Сеченова» Минздрава России (Сеченовский Университет)
27. Скворцова Тамара Андреевна, к.м.н., Главный внештатный детский специалист гастроэнтеролог Департамента здравоохранения г. Москва, руководитель Центра детской гастроэнтерологии и Центра ВЗК, заведующая отделением гастроэнтерологии ГБУЗ «Морозовская детская городская клиническая больница ДЗМ»
28. Строкова Татьяна Викторовна, д.м.н., профессор РАН, заведующая отделением педиатрической гастроэнтерологии, гепатологии и диетотерапии Клиники ФГБУН "ФИЦ питания и биотехнологии"
29. Субботин Дмитрий Михайлович, врач-генетик ФГБНУ «Медико-генетический научный центр им. академика Н.П. Бочкова», член Ассоциации медицинских генетиков.
30. Сурков Андрей Николаевич, д.м.н., заведующий гастроэнтерологическим отделением с гепатологической группой ФГАУ «НМИЦ здоровья детей» Минздрава России
31. Туманова Елена Леонидовна, д.м.н., профессор, заведующая кафедрой патологической анатомии ГБОУ ВПО «РНИМУ им. Н.И. Пирогова Минздрава России»
32. Цимбалова Екатерина Георгиевна, к.м.н., внештатный специалист МЗ МО-главный детский гастроэнтеролог МО, член Союза педиатров России, член детской группы Российского общества по изучению ВЗК.
Представитель пациентских сообществ:
33. Погосян Нелли Сергеевна – представитель Всероссийского общества орфанных заболеваний
Авторы подтверждают отсутствие финансовой поддержки/конфликта интересов, который необходимо обнародовать.
Приложение А2. Методология разработки клинических рекомендаций
Настоящие рекомендации предназначены для применения медицинскими организациями и учреждениями федеральных, территориальных и муниципальных органов управления здравоохранением, систем обязательного и добровольного медицинского страхования, другими медицинскими организациями различных организационно-правовых форм деятельности, направленной на оказание медицинской помощи.
Настоящие рекомендации устанавливают виды, объем и индикаторы качества медицинской помощи пациентам при ДЛКЛ и были рассмотрены 1-2 июня 2018 года в рамках Научно-практического конгресса «Орфанные болезни» в Москве.
Клинические рекомендации созданы на основании систематического обзора литературы 1992-2019 гг. Medline (Pubmed version), Embase (Dialog version) и Cochrane Library databases современных международных клинических рекомендаций по диагностике, лечению и ведению пациентов с метаболическими болезнями.
Дефицит лизосомной кислой липазы относятся к редким наследственным заболеваниям, что исключает возможность проведения больших когортных и рандомизированных контролированных исследований и для создания протоколов диагностики и терапии используются лишь тематические исследования экспертов, опубликованные в последние два десятилетия.
Оценка качества доказательств и силы рекомендаций применения медицинских технологий проводилась в соответствии с унифицированной шкалой, приведенной в таблицах 1-3.
Целевая аудитория данных клинических рекомендаций:
1. Врачи общей врачебной практики (семейные врачи);
2. Врачи-педиатры;
3. Врачи-гастроэнтерологи;
4. Врачи-терапевты;
5. Врачи-генетики;
6. Врачи-лабораторные генетики;
7. Врачи-кардиологи;
8. Врачи-детские кардиологи;
9. Врачи-неврологи;
10. Врачи-рентгенологи;
11. Врачи функциональной диагностики;
12. Медицинские психологи;
13. Студенты медицинских ВУЗов;
14. Обучающиеся в ординатуре.
Таблица 1. Шкала оценки уровней достоверности доказательств (УДД) для методов диагностики (диагностических вмешательств)
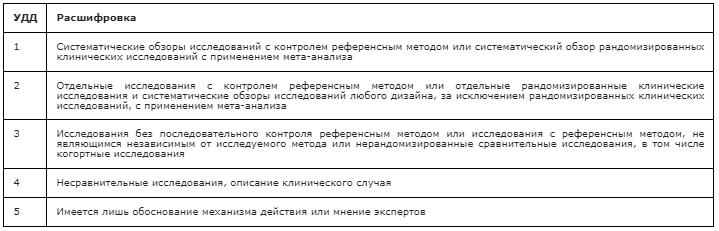
Таблица 2. Шкала оценки уровней достоверности доказательств (УДД) для методов профилактики, лечения и реабилитации (профилактических, лечебных, реабилитационных вмешательств)

Таблица 3. Шкала оценки уровней убедительности рекомендаций (УУР) для методов профилактики, диагностики, лечения и реабилитации (профилактических, диагностических, лечебных, реабилитационных вмешательств)

Порядок обновления клинических рекомендаций.
Механизм обновления клинических рекомендаций предусматривает их систематическую актуализацию – не реже чем один раз в три года, а также при появлении новых данных с позиции доказательной медицины по вопросам диагностики, лечения, профилактики и реабилитации конкретных заболеваний, наличии обоснованных дополнений/замечаний к ранее утверждённым КР, но не чаще 1 раза в 6 месяцев.
Приложение А3. Справочные материалы, включая соответствие показаний к применению и противопоказаний, способов применения и доз лекарственных препаратов, инструкции по применению лекарственного препарата
-
Федеральный закон "Об основах охраны здоровья граждан в Российской Федерации" (N 323-ФЗ от 21.11.2011).
-
Приказ Минздрава России от 21.04.2022 N 274н "Об утверждении Порядка оказания медицинской помощи пациентам с врожденными и (или) наследственными заболеваниями".
-
Приказ Министерства здравоохранения и социального развития РФ от 16 апреля 2012 г. N 366н "Об утверждении Порядка оказания педиатрической помощи"
-
Приказ Министерства здравоохранения Российской Федерации от 12 ноября 2012 г. № 906н "Об утверждении Порядка оказания медицинской помощи населению по профилю "гастроэнтерология"
Информация о лекарственных средствах: https://grls.rosminzdrav.ru/
Основные нормативно-правовые акты, регулирующие оказание паллиативной медицинской помощи
-
Федеральный закон "О внесении изменений в Федеральный закон "Об основах охраны здоровья граждан в Российской Федерации" по вопросам оказания паллиативной медицинской помощи" от 06.03.2019 № 18-ФЗ.
-
Приказ Минздрава России № 345н, Минтруда России от 31.05.2019 № 372н «Об утверждении положения об организации оказания паллиативной медицинской помощи, включая порядок взаимодействия медицинских организаций, организаций социального обслуживания и общественных объединений, иных некоммерческих организаций, осуществляющих свою деятельность в сфере охраны здоровья».
-
Приказ Минздрава России № 348н от 31 мая 2019 года «Об утверждении перечня медицинских изделий, предназначенных для поддержания органов и систем организма человека, предоставляемых для использования на дому».
-
Приказ Минздрава России № 505н от 10 июля 2019 года «Об утверждении Порядка передачи от медицинской организации пациенту (его законному представителю) медицинских изделий, предназначенных для поддержания функций органов и систем организма человека, для использования на дому при оказании паллиативной медицинской помощи».
Прочие нормативно-правовые документы, с учетом которых разработаны клинические рекомендации:
-
Федеральный закон от 21 ноября 2011 г. № 323-ФЗ «Об основах охраны здоровья граждан в Российской Федерации» (Собрание законодательства Российской Федерации, 2011 г., № 48, ст. 6724);
-
Международная классификация болезней, травм и состояний, влияющих на здоровье (МКБ – 10);
-
Приказ МЗ РФ от 20 декабря 2012г. № 1183н «Об утверждении номенклатуры должностей медицинских работников и фармацевтических работников»;
-
Приказ Минздрава России № 103н от 28.02.2019 г. «Об утверждении порядка и сроков разработки клинических рекомендаций, их пересмотра, типовой формы клинических рекомендаций и требований к их структуре, составу и научной обоснованности включаемой в клинические рекомендации информации».
-
Приказ Минздрава России от 13.10.2017 N 804н "Об утверждении номенклатуры медицинских услуг".
-
Приказ Министерства труда и социальной защиты РФ от 27 августа 2019 г. №585н "О классификациях и критериях, используемых при осуществлении медико-социальной экспертизы граждан федеральными государственными учреждениями медико-социальной экспертизы";
Сравнение проявлений инфантильной формы ДЛКЛ (болезни Вольмана) и болезни накопления эфиров холестерина (БНЭХ)

Дифференциальная диагностика НАЖБП и ДЛКЛ
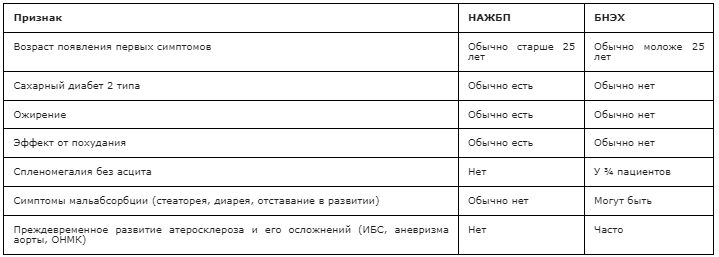
Забор биоматериала для диагностики в пятнах крови

Алгоритм действий медицинского персонала при взятии образцов крови
· вымыть руки (гигиенический уровень), надеть перчатки;
· вымыть руки пациента (пятку ребенка в случае, если кровь берется из пятки);
· протереть область прокалывания стерильной салфеткой, смоченной 70% спиртом, промокнуть сухой стерильной салфеткой; - проколоть стерильным одноразовым скарификатором;
· снять первую каплю крови стерильным сухим тампоном;
· мягко надавить для получения второй капли крови;
· приложить перпендикулярно тест-бланк к капле крови и пропитать его кровью насквозь;
· аналогичным образом нанести на тест-бланк 6-8 капель, вид пятен крови должен быть одинаковым с обеих сторон.
· высушить тест-бланк в горизонтальном положении на чистой обезжиренной поверхности не менее 4 ч без применения тепловой обработки и попадания прямых солнечных лучей;
· упаковать тест-бланки в чистый конверт таким образом, чтобы пятна крови не соприкасались.
Особенности при инфузионной терапии
Некоторые пациенты могут получать инфузионную терапию, переливание компонентов крови, что может оказать влияние на результаты тестов. Например, при переливании плазмы крови могут быть получены ложноотрицательные результаты, так как определяемые ферменты находятся в плазме и в клетках крови. Рекомендуется осуществить забор крови для ферментной и ДНК-диагностики не 9 ранее чем через 6-7 дней после переливания плазмы крови и через 7-10 дней после переливания компонентов крови
Не допускается забор крови
· сразу после проведения пациенту инфузионной терапии;
· сразу после заменного переливания крови.
Хранение и транспортировка биоматериала
Образцы высушенных пятен крови можно хранить в обычной камере холодильника при +40С до отправки. Срок хранения до момента отправки не должен превышать 7 дней. Если хранить дольше и при более высокой температуре, то активность фермента даже в норме может снижаться, что приведет к ложноположительным результатам.
Рекомендованные объемы введения себелипазы альфа**
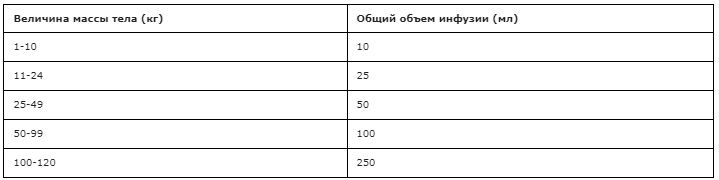
* Объем инфузии должен базироваться на назначенной дозе и должен быть приготовлен до окончательной концентрации себелипазы альфа** 0,1‑1,5 мг/мл.
(доза 1 мг/кг) (https://grls.rosminzdrav.ru/)
Расшифровка примечаний
…** – препарат входит в перечень жизненно необходимых и важнейших лекарственных препаратов (Распоряжение Правительства РФ от 12.10.2019 № 2406-р «Об утверждении перечня жизненно необходимых и важнейших лекарственных препаратов на 2020 год, а также перечней лекарственных препаратов для медицинского применения и минимального ассортимента лекарственных препаратов, необходимых для оказания медицинской помощи»
# - применение off-label – вне зарегистрированных в инструкции лекарственного средства показаний осуществляется по решению врачебной комиссии, с разрешения Локального этического комитета медицинской организации (при наличии), с условием подписанного информированного согласия родителей (законного представителя) и пациента в возрасте старше 15 лет;
*** - медицинское изделие, имплантируемое в организм человека при оказании медицинской помощи в рамках программы государственных гарантий бесплатного оказания гражданам медицинской помощи.
Приложение Б. Алгоритмы действий врача
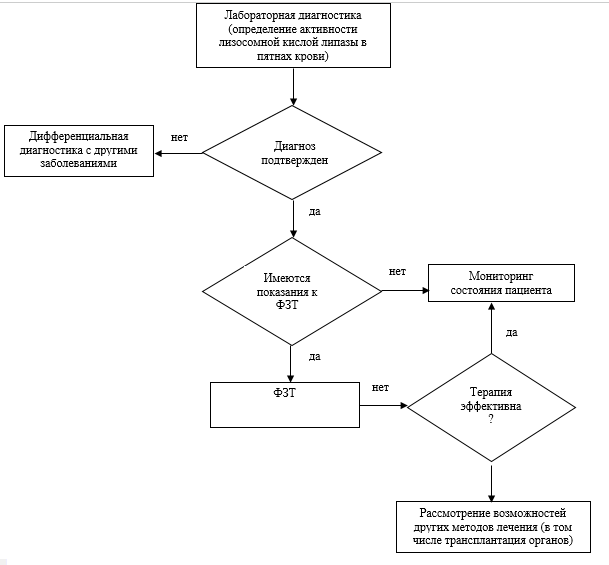
Приложение В. Информация для пациента
Почему возникает заболевание?
ДЛКЛ — наследственное заболевание и связано с поломками (мутациями) в гене LIPA.
Гены представляют собой последовательность ДНК и в них записаны инструкции по «приготовлению» белков или РНК (рибонуклеиновая кислота). Гены находятся в хромосомах. У человека 23 пары хромосом, одну из хромосом с соответствующим набором генов он наследует от матери, вторую от отца. Среди хромосом еще есть две особые хромосомы, которые определяют пол ребенка — у девочек две Х-хромосомы, а у мальчиков одна Х и одна У-хромосомы.
Ген LIPA находится на 10 хромосоме.
ДЛКЛ наследуется по аутосомно-рецессивному типу, что означает, что оба родителя являются носителями мутации, но не болеют, т.к. у них есть вторая копия здорового гена. Ребенок с ДЛКЛ наследует одну копию “больного” гена от отца и одну— от матери. Заболевание проявляется только в случае наличия двух копий поврежденного гена. Риск рождения ребенка с ДЛКЛ в данном случае составляет 25%. Мальчики и девочки болеют с одинаковой частотой. Носитель болезни наследует только одну копию либо от отца, либо от матери. Они не болеют и никаких признаков болезни у них нет.
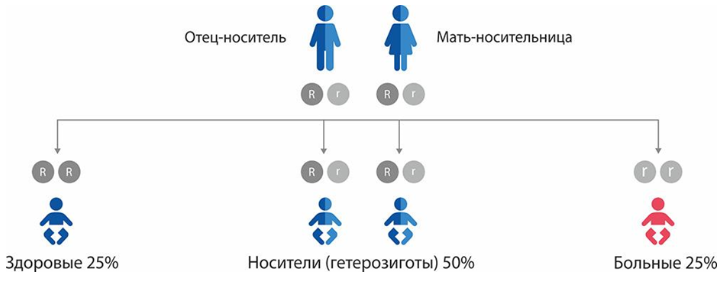
Что нарушается в метаболизме при ДЛКЛ?
При ДЛКЛ фермент лизосомная кислая липаза (ЛКЛ) не работает, что приводит к накоплению в особых клеточных органеллах, называемых лизосомами, таких крупных молекул как триглицериды и эфиры холестерина. В норме триглицериды и эфиры холестерина расщепляются в лизосомах, а образующиеся жирные кислоты и холестерин усваиваются организмом для строительства новых клеток и для пополнения энергии. У пациентов с ДЛКЛ из-за недостатка активности ЛКЛ клетки настолько переполняются нерасщеплёнными молекулами, что они перестают выполнять свои функции. Больше всего таких нефункционирующих клеток находится в печени, селезенке, стенках кровеносных сосудов, надпочечниках, но поражаются также и другие органы и ткани. Со временем, например, печень, значительно увеличивается в размерах, перестает функционировать, после чего орган зарастает соединительной тканью с переходом в цирроз. А повышение уровня холестерина в крови сопряжено с повышенным риском заболеваний сердца, а также рядом других заболеваний, вызванных нарушением проходимости артерий.
Как проявляется ДЛКЛ?
Впервые описанные М. Вольманом как отдельные заболевания, инфантильная форма ДЛКЛ (болезнь Вольмана) (в 1956 году) и болезнь накопления эфиров холестерина (в 1963 году), в настоящее время рассматриваются как клинические формы ДЛКЛ.
Возраст начала заболевания и темпы его прогрессирования в значительной степени вариабельны и могут быть связаны с природой, лежащих в основе мутаций.
При инфантильной форме ДЛКЛ (болезни Вольмана) у младенцев возникают глубокие нарушения роста и развития, фиброз печени, цирроз; дети погибают от печеночной недостаточности в раннем возрасте.
При поздней форме ДЛКЛ (БНЭХ) у детей и взрослых наблюдается сочетание дислипидемии (нарушение баланса липидов в организме), гепатомегалии (увеличение печени), повышения уровня трансаминаз (ферментов, уровень которых повышается при заболеваниях печени) и микровезикулярного стеатоза (накопления жировых капель в клетках печени).
У детей и взрослых с ДЛКЛ на протяжении 3 лет после начала симптомов приблизительно в 50 % случаев наблюдается прогрессирование в фиброз, цирроз или возникает необходимость в проведении трансплантации печени. Начиная с детского возраста, могут проявляться нарушения со стороны сердечно-сосудистой системы, обусловленные ранним атеросклерозом.
Диагноз ДЛКЛ может быть заподозрен при наличии изменений со стороны печени (гепатомегалии, повышения активности трансаминаз, признаков стеатоза) и нарушения липидного профиля. Сложность диагностики заключается в отсутствии или немногочисленности жалоб пациента, несмотря на прогрессирование патологического процесса в печени и других органах. Во многих случаях ДЛКЛ находят случайно при обнаружении изменений в анализе крови биохимическом общетерапевтическом или ультразвуковом исследовании печени. Тем не менее, различные органы, в большей степени печень, теряют свою функциональность. В связи с тем, что организм может некоторое время функционировать даже с небольшой частью печени, некоторые пациенты с ДЛКЛ узнают о своем диагнозе, имея последние стадии цирроза, когда любой стрессовое состояние может закончиться полной потерей функционирования печени и даже смертью пациента.
Со стороны печени:
• Фиброз
• Цирроз
• Портальная гипертензия
• Печеночная недостаточность
Со стороны сердечно-сосудистой системы:
• Ускоренное формирование атеросклероза
• Ишемическая болезнь сердца
• Инсульт
• Инфаркт миокарда
Со стороны селезенки:
• Анемия
• Тромбоцитопения
• Риск травматического разрыва селезенки и/или спленэктомии
Со стороны желудочно-кишечного тракта
• Боль в животе
• Нарушение пищеварения
• Отставание в росте
Какие у этого заболевания бывают осложнения?
По мере прогрессирования ДЛКЛ может развиваться печеночная недостаточность. В связи с нарушением обмена жиров и холестерина может развиться атеросклероз, что приводит к инфарктам и инсультам. Анемия и снижение количества тромбоцитов, а значит и частые кровотечения – это осложнение ДЛКЛ в связи с нарушением функции селезенки.
Как устанавливают в лаборатории диагноз ДЛКЛ?
Врачи на основании клинических симптомов могут заподозрить болезнь. Затем проводятся лабораторные тесты и инструментальное исследование. Диагноз ДЛКЛ подтверждается на основании обнаружения значительного дефицита активности фермента. Активность ЛКЛ оценивается путем определения активности фермента в сухих пятнах крови на фильтрах. Такой способ определения практически однозначно указывает на ДЛКЛ, так как у пациентов активность ЛКЛ снижена в десятки и сотни раз, а иногда, при тяжелых формах, вовсе отсутствует. Неизвестно больше никаких других состояний, когда активность ЛКЛ настолько низкая. Анализ крови на ЛКЛ может выступать в качестве инструмента в программах скрининга и крупных популяционных исследованиях ДЛКЛ, а также может быть адаптирован для скрининга новорожденных. Если активность фермента снижена, то проводится ДНК-диагностика. В результате которой выявляют мутации в гене LIPA. При этом заболевании у пациента выявляют две мутации в гене. Они могут быть в гомозиготном состоянии (то есть две одинаковые мутации) или в компаунд-гетерозиготном состоянии (две разные мутации).
Только на основании лабораторного тестирования диагноз может быть установлен.
Какой прогноз у пациентов с ДЛКЛ?
Без лечения прогноз неблагоприятный. Младенцы с инфантильной формой ДЛКЛ (болезнью Вольмана) погибают в течение первых шести месяцев жизни.
У половины детей и взрослых с медленно развивающейся формой болезни (болезнь накопления эфиров холестерина) в течение 3х лет после появления симптомов ДЛКЛ поражение печени становится настолько тяжелым, что развивается фиброз, цирроз или требуется пересадка печени.
Как лечат пациентов с ДЛКЛ?
Для лечения недавно существует препарат, который является ферментом липазой, но только полученной генно-инженерным путем. Это препарат называется «Себелипаза альфа**» и он уже зарегистрирован в России. Суть лечения очень простая – пациенту вводят недостающий в организме фермент, в результате себелипаза альфа** начинает расщеплять накапливаемые вещества и останавливает прогрессирование болезни.
Терапия является пожизненной или можно пройти несколько курсов и вылечить болезнь?
Заместительная ферментная терапия себелипазой альфа** является пожизненной и значительно улучшает прогноз заболевания, улучшает качество жизни детей с ДЛКЛ и предотвращает развитие цирроза печени. К сожалению, фермент, который вводят внутривенно «живет» только ограниченное время и его недостаток нужно все время восполнять.
Себелипаза альфа** назначается 1раз в две недели детям и взрослым и 1 раз в неделю младенцам. Доза составляет 1мг/кг, то есть рассчитывается по весу ребенка. Препарат вводится внутривенно. Коррекция дозы препарата проводится при снижении или увеличении веса ребенка.
Чего стоит опасаться при назначении себелипазы альфа**?
3% пациентов во время клинических исследований указывали на признаки и симптомы анафилаксии, то есть тяжелой аллергической реакцией. Поэтому инфузии себелипазы альфа** должны проводиться подготовленным медицинским работником, умеющим бороться с тяжелыми аллергическими реакциями. В случае развития подобных реакций, необходимо незамедлительно прекратить введение препарата и начать соответствующее лечение.
Можно ли лечить заболевание другими препаратами — которые снижают уровень холестерина или помогают работе печени?
Эта терапия, которая применялась до появления препарата, способного воздействовать на течение болезни. К сожалению, такое лечение является симптоматическим, имеет нестойкий и непродолжительный эффект, не влияет на исходы заболевания. Кроме того, исследований, показывающих эффективность этой терапии не проводилось. Однако надо отметить, что один из подходов к терапии сохраняет актуальность до сих пор и улучшает состояние пациентов с ДЛКЛ. Это низкожировая диета. Однако без проведения заместительной ферментной терапии ее эффективность будет недостаточной.
Таблица 1. Обследование пациентов с ДЛКЛ
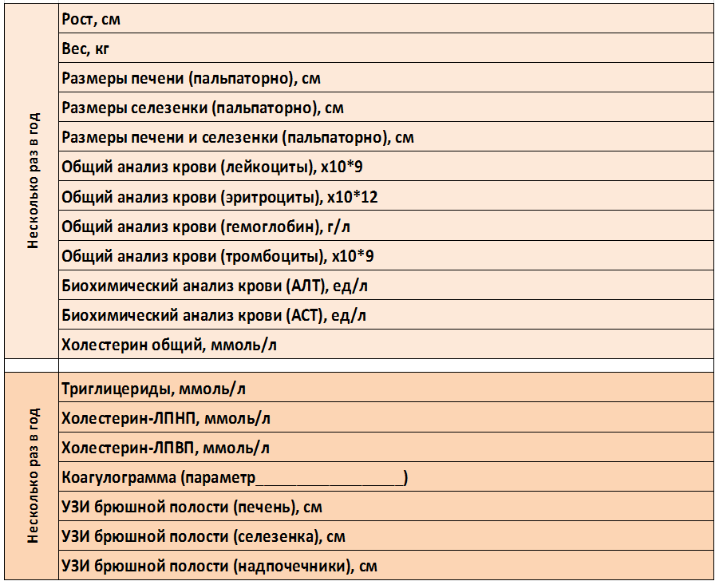
Кроме того, нужно проходить контрольное обследование в круглосуточном или дневном стационаре 2 раза в год для оценки эффективности состояния.
Приложение Г1-ГN. Шкалы оценки, вопросники и другие оценочные инструменты состояния пациента, приведенные в клинических рекомендациях
Не применимо.
Прикреплённые файлы
Внимание!
- Занимаясь самолечением, вы можете нанести непоправимый вред своему здоровью.
- Информация, размещенная на сайте MedElement и в мобильных приложениях "MedElement (МедЭлемент)", "Lekar Pro", "Dariger Pro", "Заболевания: справочник терапевта", не может и не должна заменять очную консультацию врача. Обязательно обращайтесь в медицинские учреждения при наличии каких-либо заболеваний или беспокоящих вас симптомов.
- Выбор лекарственных средств и их дозировки, должен быть оговорен со специалистом. Только врач может назначить нужное лекарство и его дозировку с учетом заболевания и состояния организма больного.
- Сайт MedElement и мобильные приложения "MedElement (МедЭлемент)", "Lekar Pro", "Dariger Pro", "Заболевания: справочник терапевта" являются исключительно информационно-справочными ресурсами. Информация, размещенная на данном сайте, не должна использоваться для самовольного изменения предписаний врача.
- Редакция MedElement не несет ответственности за какой-либо ущерб здоровью или материальный ущерб, возникший в результате использования данного сайта.