Диагностика и лечение пациентов с наследственными нервно-мышечными заболеваниями (взрослое население)
![]() Версия: Клинические протоколы 2025 (Беларусь)
Версия: Клинические протоколы 2025 (Беларусь)
Болезни накопления гликогена (E74.0), Наследственная моторная и сенсорная невропатия (G60.0), Первичные поражения мышц (G71), Спинальная мышечная атрофия и родственные синдромы (G12)
Медицинская реабилитация, Неврология
Общая информация
Краткое описание
УТВЕРЖДЕНО
Постановление
Министерства здравоохранения
Республики Беларусь
13.05.2025 № 42
КЛИНИЧЕСКИЙ ПРОТОКОЛ
«Диагностика и лечение пациентов с наследственными нервно-мышечными заболеваниями (взрослое население)»
ГЛАВА 1
ОБЩИЕ ПОЛОЖЕНИЯ
1. Настоящий клинический протокол устанавливает общие требования к оказанию медицинской помощи пациентам (взрослое население) с наследственными нервно-мышечными заболеваниями (далее – ННМЗ) (шифр по Международной статистической классификации болезней и проблем, связанных со здоровьем, десятого пересмотра – G12 Спинальная мышечная атрофия и родственные синдромы, G60.0 Наследственная моторная и сенсорная невропатия, G71 Первичные поражения мышц, Е74.0 Болезни накопления гликогена).
2. Требования настоящего клинического протокола являются обязательными для юридических лиц и индивидуальных предпринимателей, осуществляющих медицинскую деятельность в порядке, установленном законодательством о здравоохранении.
3. Для целей настоящего клинического протокола используются основные термины и их определения в значениях, установленных Законом Республики Беларусь «О здравоохранении».
4. Настоящий клинический протокол определяет минимальный объем медицинской помощи пациентам с ННМЗ при оказании специализированной медицинской помощи врачами-неврологами, врачами-генетиками, врачами-реабилитологами, врачами-травматологами-ортопедами, врачами-пульмонологами, врачами-гастроэнтерологами и (или) врачами-диетологами, врачами общей практики, врачами-терапевтами, врачами-специалистами службы паллиативной медицинской помощи и другими врачами-специалистами с учетом медицинских показаний в конкретном случае.
5. Для лечения ННМЗ рекомендованы базовые схемы медикаментозного лечения заболеваний, включающие основные группы лекарственных препаратов (далее – ЛП).
ЛП представлены в главе 4 настоящего клинического протокола в соответствии с международными непатентованными наименованиями, а при их отсутствии – по химическим наименованиям по систематической или заместительной номенклатуре с указанием пути введения, лекарственных форм и дозировок, режима дозирования и разовой (при необходимости суточной, максимальной разовой) дозы.
Применение ЛП осуществляется по медицинским показаниям в соответствии с общей характеристикой ЛП. Допускается включение в схему лечения ЛП по медицинским показаниям или в режиме дозирования, не утвержденным инструкцией по медицинскому применению (листком-вкладышем) и общей характеристикой ЛП (off-label), с дополнительным обоснованием и указанием особых условий назначения, способа применения, дозы, длительности и кратности приема. Дополнительно указываются особые условия назначения, способ применения, доза, длительность и кратность приема.
6. В каждой конкретной ситуации в интересах пациента при наличии медицинских показаний (по жизненным показаниям, с учетом индивидуальной непереносимости и (или) чувствительности) по решению врачебного консилиума объем диагностики и лечения может быть расширен с использованием других методов, не включенных в настоящий клинический протокол.
7. Координация работы врачей-специалистов и контроль оказания медицинской помощи осуществляются врачом-неврологом на основании заключения врачебного консилиума, проведенного в республиканском центре ННМЗ у пациентов старше 18 лет (далее – Центр) на базе государственного учреждения «Республиканский научно-практический центр неврологии и нейрохирургии» (далее – РНПЦ неврологии и нейрохирургии), с участием руководителя Центра (врача-невролога) и врачей-специалистов по медицинским показаниям.
Классификация
ГЛАВА 2
КЛАССИФИКАЦИЯ И КЛИНИЧЕСКИЕ ПРОЯВЛЕНИЯ ННМЗ
8. ННМЗ представляют многочисленную группу генетически гетерогенных наследственных болезней нервной системы, в основе которых лежит поражение передних рогов спинного мозга, периферических нервов и скелетных мышц, приводящих к выраженному клиническому полиморфизму.
9. Термин «спинальная мышечная атрофия» (далее – СМА) используется в настоящем клиническом протоколе в значении, определенном абзацем вторым пункта 3 клинического протокола «Оказание медицинской помощи пациентам детского возраста со спинальной мышечной атрофией», утвержденного постановлением Министерства здравоохранения Республики Беларусь от 5 мая 2023 г. № 73.
Термин «генетическая причина СМА» используется в настоящем клиническом протоколе в значении, определенном пунктом 4 клинического протокола «Оказание медицинской помощи пациентам детского возраста со спинальной мышечной атрофией».
На основании возраста проявления клинических симптомов заболевания, максимальной достигаемой двигательной функции с последующим ее регрессом выделяют 5 типов СМА, представленных в пункте 6 клинического протокола «Оказание медицинской помощи пациентам детского возраста со спинальной мышечной атрофией».
10. Бульбоспинальная атрофия Кеннеди (экспансия CAG-тринуклеотидных повторов в гене AR, располагающемся на Х-хромосоме и кодирующем андрогеновый рецептор).
Наследуется по Х-сцепленному рецессивному типу. Возраст дебюта заболевания – от 27 до 43 лет. Клинические проявления: атрофия и мышечная слабость в дистальных и проксимальных отделах конечностей, нарушение глотания, речи, походки, контрактуры суставов, крампи, развитие аспирационной пневмонии и дыхательной недостаточности на фоне слабости дыхательной мускулатуры, гастроэзофагеальный рефлюкс, запоры, снижение чувствительности к андрогенам (гинекомастия, атрофия яичек, олиго или азооспермия).
11. Наследственная моторная и сенсорная невропатия (далее – НМСН) – гетерогенная группа генетически детерминированных заболеваний, в основе которых лежит первичный дефект в структуре периферических нервов (миелиновой оболочке, аксоне), различающихся по типу наследования (аутосомно-доминантный, аутосомно-рецессивный, X-сцепленный), клиническому полиморфизму, особенностям электронейромиографических (далее – ЭНМГ) параметров:
- НМСН 1 типа (мутации в генах РМР22, MPZ, GJB1, EGR2, LITAF;
- демиелинизирующий вариант). Возраст дебюта заболевания – первое десятилетие жизни, реже – в начале второго. Клинические проявления: слабость и деформации стоп, медленно прогрессирующая дистальная мышечная атрофия («ноги аиста»), спазмы мышц голеней, изменение походки, затруднения при беге и (или) подъеме по лестнице. Позднее присоединяются атрофии мышц и слабость в кистях. Вибрационная, температурная и болевая чувствительность нарушаются по типу «перчаток» и «носков». Выпадают глубокие рефлексы;
- НМСН II типа (мутации в генах MFN2, RAB7; аксональный вариант). Возраст дебюта заболевания – от 30 до 60 лет. Клинические проявления: снижение силы, атрофии в дистальных отделах конечностей, деформации стоп, вегетативные и трофические нарушения, нейропатический болевой синдром, медленно-прогрессирующее течение;
Возраст дебюта заболевания – первые 2 года жизни. Клинические проявления:
12. Первичные поражения мышц – гетерогенная группа заболеваний с аутосомно-доминантным, аутосомно-рецессивным и Х-сцепленным типом наследования, проявляющихся прогрессирующей мышечной слабостью вследствие первичного поражения мышц:
12.1. мышечные дистрофии (далее – МД):
12.1.1. МД Дюшенна (мутации в гене DMD, рецессивный тип наследования, сцепленный с Х-хромосомой). Возраст дебюта заболевания – 2–5 лет. Клинические проявления: прогрессирующая мышечная слабость, атрофии мышц тазового, плечевого пояса, проксимальных отделов конечностей, псевдогипертрофия икроножных мышц, нарушение осанки, контрактуры суставов, дыхательные нарушения, кардиомиопатия, снижение интеллекта. Средняя продолжительность жизни 25 лет;
12.1.2. МД Беккера (мутации в гене DMD, рецессивный тип наследования, сцепленный с Х хромосомой). Возраст дебюта заболевания – 5–10 лет. Клинические проявления: медленно прогрессирующая мышечная слабость, отсутствие контрактур, кардиомиопатия, сохранный интеллект, сохранение способности к самостоятельной ходьбе в течение 15–20 лет от начала заболевания;
12.1.3. окулофарингеальная МД (мутации в гене PABPN1).
12.1.4. МД Эмери-Дрейфуса (мутации в гене EMD, FNL1, LMNA, тип наследования аутосомно-рецессивный, сцепленный с Х-хромосомой, реже аутосомно-доминантный).
- НМСН III типа (мутации в генах РМР22, P0, EGR2; болезнь Дежерина-Сотта).
Возраст дебюта заболевания – первые 2 года жизни. Клинические проявления:
- выраженный дистальный тетрапарез, расстройство чувствительности, сколиоз, контрактуры суставов, дыхательные расстройства, нарушение глотания, страбизм и офтальмоплегия;
- НМСН IV типа (мутации в гене GDAP1, болезнь Рефсума). Возраст дебюта заболевания – первые 2 года жизни. Клинические проявления: прогрессирующая атрофия мышц дистальных отделов рук и ног, нарушение походки типа «степпаж», снижение поверхностной чувствительности по полиневритическому типу, отсутствие сухожильных рефлексов, тяжелое злокачественное течение;
- НМСН V типа (мутации в гене NGFB). Возраст дебюта заболевания – 2–3 десятилетие жизни. Клинические проявления: слабость в дистальных отделах рук и ног с развитием выраженной атрофии, деформации стоп: характерно сочетание симптомов моторной полиневропатии с пирамидной симптоматикой;
- НМСН VI типа (мутации в гене MFN2). Возраст дебюта заболевания – от 6 месяцев до 50 лет. Клинические проявления: сочетание симптомов периферической невропатии, атрофии зрительных нервов и нейросенсорной тугоухости;
- НМСН VIIA типа (мутации в гене SLC5A7). Возраст дебюта заболевания – от рождения до 16 лет. Клинические проявления: пигментная дегенерация сетчатки, бульбарные нарушения, паралич голосовых складок, слабость лицевой и аксиальной мускулатуры, проксимальных отделов конечностей, нарушение интеллекта;
- НМСН VIIB типа (мутации в гене DCTN1). Возраст дебюта заболевания – от 3 до 35 лет. Клинические проявления: пигментная дегенерация сетчатки, прогрессирующая слабость лицевой мускулатуры, паралич голосовых складок, мышечная слабость в дистальных отделах конечностей, контрактуры голеностопных суставов.
12. Первичные поражения мышц – гетерогенная группа заболеваний с аутосомно-доминантным, аутосомно-рецессивным и Х-сцепленным типом наследования, проявляющихся прогрессирующей мышечной слабостью вследствие первичного поражения мышц:
12.1. мышечные дистрофии (далее – МД):
12.1.1. МД Дюшенна (мутации в гене DMD, рецессивный тип наследования, сцепленный с Х-хромосомой). Возраст дебюта заболевания – 2–5 лет. Клинические проявления: прогрессирующая мышечная слабость, атрофии мышц тазового, плечевого пояса, проксимальных отделов конечностей, псевдогипертрофия икроножных мышц, нарушение осанки, контрактуры суставов, дыхательные нарушения, кардиомиопатия, снижение интеллекта. Средняя продолжительность жизни 25 лет;
12.1.2. МД Беккера (мутации в гене DMD, рецессивный тип наследования, сцепленный с Х хромосомой). Возраст дебюта заболевания – 5–10 лет. Клинические проявления: медленно прогрессирующая мышечная слабость, отсутствие контрактур, кардиомиопатия, сохранный интеллект, сохранение способности к самостоятельной ходьбе в течение 15–20 лет от начала заболевания;
12.1.3. окулофарингеальная МД (мутации в гене PABPN1).
Аутосомно-доминантный тип наследования: возраст дебюта заболевания – 40–60 лет. Клинические проявления: птоз, слабость круговых мышц глаз и рта, дисфагия, атрофия и слабость мышц плечевого и тазового пояса.
Аутосомно-рецессивный тип наследования: возраст дебюта заболевания – от 40–50 лет. Клинические проявления: характерно вовлечение дистальных групп мышц конечностей, помимо проксимальной мускулатуры;
12.1.4. МД Эмери-Дрейфуса (мутации в гене EMD, FNL1, LMNA, тип наследования аутосомно-рецессивный, сцепленный с Х-хромосомой, реже аутосомно-доминантный).
Возраст дебюта заболевания – с рождения и до 30 лет. Клинические проявления: медленно прогрессирующая слабость лопаточно-плечевой и тазово-перинеальной групп мышц, кардиомиопатия; быстро развивающиеся контрактуры локтевых и голеностопных суставов;
12.2. миотонические расстройства:
12.2.1. миотония Томсена, мутации в гене хлорного канала CLCN1, локус 7q35; аутосомно-доминантный тип наследования. Возраст дебюта заболевания – первые 3 года жизни. Клинические проявления: генерализованные миотонические феномены, слабость в мышцах верхних и нижних конечностей, лицевой мускулатуре, гипертрофия скелетной мускулатуры, миалгии (редко);
12.2.2. миотония Беккера, мутации в гене хлорного канала CLCN1, локус 7q35; аутосомно-рецессивный тип наследования. Возраст дебюта заболевания – 8–10 лет.
12.2.3. дистрофическая миотония, 1 тип: увеличение числа триплетов CTG в 3’-нетранслируемой области гена DMPK на хромосоме 19; 2 тип: возрастание количества повторов CCTG в интроне 1 гена CNBP (ZNF9) на хромосоме 3; аутосомно-доминантный тип наследования). Возраст дебюта заболевания – с рождения до 70 лет.
12.3. врожденные миопатии:
12.3.1. болезнь центрального стержня, связанная со злокачественной гипертермией.
12.3.2. немалиновая миопатия с аутосомно-доминантным (мутации в гене ACTA-1) и аутосомно-рецессивным (мутации в гене NEB) типом наследования. Возраст дебюта заболевания – с рождения до 55 лет. Клинические проявления: характерный внешний вид пациента (арахнодактилия, удлиненное лицо, деформация грудной клетки), мышечная гипотония, генерализованная мышечная слабость, кифосколиоз, концентрация КФК может повышаться;
12.4. митохондриальные миопатии:
12.4.1. синдром MERRF, миоклоническая эпилепсия с разорванными красными волокнами (точечная мутация в гене лизиновой транспортной РНК). Возраст дебюта заболевания – от 3 до 65 лет. Клинические проявления: мышечная слабость, судорожные приступы (атонические, тонико-клонические, миоклонические), атаксия, деменция;
12.4.2. синдром MELAS, митохондриальная энцефаломиопатия с лактат-ацидозом и инсультоподобными эпизодами (точечная мутация митохондриальной РНК). Возраст дебюта заболевания – 6–10 лет. Клинические проявления: судороги, повторные эпизоды головной боли, рвота, непереносимость физических нагрузок, мышечная слабость, крампи, повторные инсульты, умственная отсталость, нейросенсорная тугоухость, офтальмоплегия, нарушение сердечной проводимости;
12.5. метаболические миопатии:
Детская форма: возраст дебюта заболевания – 1–3 года. Клинические проявления: медленно прогрессирующая мышечная слабость в проксимальных отделах конечностей, органомегалия, поражение миокарда, слабость диафрагмы и межреберных мышц, приводящие к дыхательной недостаточности.
Взрослая форма: возраст дебюта заболевания – 20–40 лет. Клинические проявления: медленно прогрессирующая мышечная слабость в проксимальных отделах конечностей, снижение двигательной активности, слабость паравертебральных мышц, мышц передней брюшной стенки, дыхательной мускулатуры с развитием дыхательной недостаточности.
12.1.5. конечностно-поясные МД. Описано более 30 генетических вариантов с аутосомно-доминантным и аутосомно-рецессивным типом наследования. Возраст дебюта заболевания – 2–3 десятилетия жизни. Клинические проявления: нарастающая слабость и атрофия мышц тазового и плечевого пояса, спины, живота, формирование гиперлордоза в поясничном отделе, «утиная» походка;
12.2. миотонические расстройства:
12.2.1. миотония Томсена, мутации в гене хлорного канала CLCN1, локус 7q35; аутосомно-доминантный тип наследования. Возраст дебюта заболевания – первые 3 года жизни. Клинические проявления: генерализованные миотонические феномены, слабость в мышцах верхних и нижних конечностей, лицевой мускулатуре, гипертрофия скелетной мускулатуры, миалгии (редко);
12.2.2. миотония Беккера, мутации в гене хлорного канала CLCN1, локус 7q35; аутосомно-рецессивный тип наследования. Возраст дебюта заболевания – 8–10 лет.
Клинические проявления: генерализованные миотонические феномены (более выражены, чем при миотонии Томсена), легкая слабость мускулатуры дистальных отделов конечностей, гипертрофия скелетной мускулатуры;
12.2.3. дистрофическая миотония, 1 тип: увеличение числа триплетов CTG в 3’-нетранслируемой области гена DMPK на хромосоме 19; 2 тип: возрастание количества повторов CCTG в интроне 1 гена CNBP (ZNF9) на хромосоме 3; аутосомно-доминантный тип наследования). Возраст дебюта заболевания – с рождения до 70 лет.
Клинические проявления: прогрессирующая мышечная слабость, атрофии, вялые дистальные парезы рук и ног в сочетании с миотоническими феноменами и внемышечными симптомами (катаракта, нарушение проводимости и сердечного ритма, кардиомиопатия), дыхательные нарушения, эндокринные расстройства (раннее облысение, азооспермия, нарушения менструального цикла, невынашивание беременности, сахарный диабет 1 типа);
12.3. врожденные миопатии:
12.3.1. болезнь центрального стержня, связанная со злокачественной гипертермией.
Аутосомно-доминантный тип наследования (мутации в гене RYR-1). Возраст дебюта заболевания – с рождения до 1 года. Клинические проявления: гипотония, гипоплазия скелетных мышц, мышечная слабость в проксимальных отделах конечностей (доминирует в руках), задержка моторного развития, глубокое угнетение рефлексов, врожденные подвывихи и гипермобильность суставов, концентрация креатинфосфокиназы (далее – КФК) не повышена.
Аутосомно-рецессивный тип наследования (мутации в гене MYH7): возраст дебюта заболевания – с рождения. Клинические проявления: преимущественно выраженная слабость разгибателей шеи, поражение сердечной мышцы;
Аутосомно-рецессивный тип наследования (мутации в гене MYH7): возраст дебюта заболевания – с рождения. Клинические проявления: преимущественно выраженная слабость разгибателей шеи, поражение сердечной мышцы;
12.3.2. немалиновая миопатия с аутосомно-доминантным (мутации в гене ACTA-1) и аутосомно-рецессивным (мутации в гене NEB) типом наследования. Возраст дебюта заболевания – с рождения до 55 лет. Клинические проявления: характерный внешний вид пациента (арахнодактилия, удлиненное лицо, деформация грудной клетки), мышечная гипотония, генерализованная мышечная слабость, кифосколиоз, концентрация КФК может повышаться;
12.4. митохондриальные миопатии:
12.4.1. синдром MERRF, миоклоническая эпилепсия с разорванными красными волокнами (точечная мутация в гене лизиновой транспортной РНК). Возраст дебюта заболевания – от 3 до 65 лет. Клинические проявления: мышечная слабость, судорожные приступы (атонические, тонико-клонические, миоклонические), атаксия, деменция;
12.4.2. синдром MELAS, митохондриальная энцефаломиопатия с лактат-ацидозом и инсультоподобными эпизодами (точечная мутация митохондриальной РНК). Возраст дебюта заболевания – 6–10 лет. Клинические проявления: судороги, повторные эпизоды головной боли, рвота, непереносимость физических нагрузок, мышечная слабость, крампи, повторные инсульты, умственная отсталость, нейросенсорная тугоухость, офтальмоплегия, нарушение сердечной проводимости;
12.4.3. синдром Кернса-Сейра (делеция митохондриальной ДНК). Возраст дебюта заболевания – до 20 лет. Клинические проявления: двусторонний птоз, расстройство глотания, нарушение сердечной проводимости, снижение слуха, мозжечковая атаксия, проксимальная мышечная слабость, сахарный диабет, дефицит гормона роста, гипопаратиреоз;
12.5. метаболические миопатии:
болезнь Помпе, болезнь накопления, связанная с дефицитом фермента кислой мальтазы в лизосомах (мутантный ген GAA, аутосомно-рецессивный тип наследования).
Ранняя инфантильная форма: возраст дебюта заболевания – до 2 месяцев жизни.
Клинические проявления: глубокая генерализованная мышечная гипотония при отсутствии мышечных атрофий, застойная сердечная недостаточность.
Детская форма: возраст дебюта заболевания – 1–3 года. Клинические проявления: медленно прогрессирующая мышечная слабость в проксимальных отделах конечностей, органомегалия, поражение миокарда, слабость диафрагмы и межреберных мышц, приводящие к дыхательной недостаточности.
Взрослая форма: возраст дебюта заболевания – 20–40 лет. Клинические проявления: медленно прогрессирующая мышечная слабость в проксимальных отделах конечностей, снижение двигательной активности, слабость паравертебральных мышц, мышц передней брюшной стенки, дыхательной мускулатуры с развитием дыхательной недостаточности.
Диагностика
ГЛАВА 3
ДИАГНОСТИКА ННМЗ
13. Обязательные диагностические исследования при ННМЗ:
13.1. клинические диагностические исследования ННМЗ:
- сбор жалоб и анамнеза, в том числе семейно-наследственного (наличие мышечной слабости, похудения, подергивания мышц, болей, судорожных сведений в мышцах, нарушения походки, чувствительности, искривления позвоночника, деформации суставов, одышки);
- медицинский осмотр, включая неврологический:
оценка состояния мышечной системы (в т.ч. наличие атрофии или гипертрофии мышц, фасцикуляций, болезненности при пальпации, миотонической реакции, снижения мышечной силы);
оценка состояния глубоких и поверхностных рефлексов, поверхностной и глубокой чувствительности, координаторной сферы, походки, функции тазовых органов;
13.2. лабораторные диагностические исследования:
- общий анализ крови;
- общий анализ мочи;
- биохимический анализ крови: креатинин, мочевина, билирубин, аланинаминотрансфераза, аспартатаминотрансфераза, щелочная фосфатаза, гаммаглютаминтранспептидаза, лактатдегидрогеназа, глюкоза, лактат, креатинфосфокиза, С-реактивный, натрий, калий, кальций ионизированный, витамин D;
- определение уровня глюкозы крови;
- коагулограмма: активированное частичное тромбопластиновое время, международное нормализованное отношение, протромбиновое время, фибриноген;
13.3. инструментальные диагностические исследования:
- электрокардиография;
- ЭНМГ, игольчатая электромиография;
- рентгеновская денситометрия;
- спирометрия;
- пикфлуометрия;
- ночная пульсоксиметрия (по медицинским показаниям);
- ультразвуковое исследование мышц;
- ультразвуковое исследование сердца;
13.4. молекулярно-генетические диагностические исследования (медицинские показания и объем молекулярно-генетических исследований определяется врачом-генетиком):
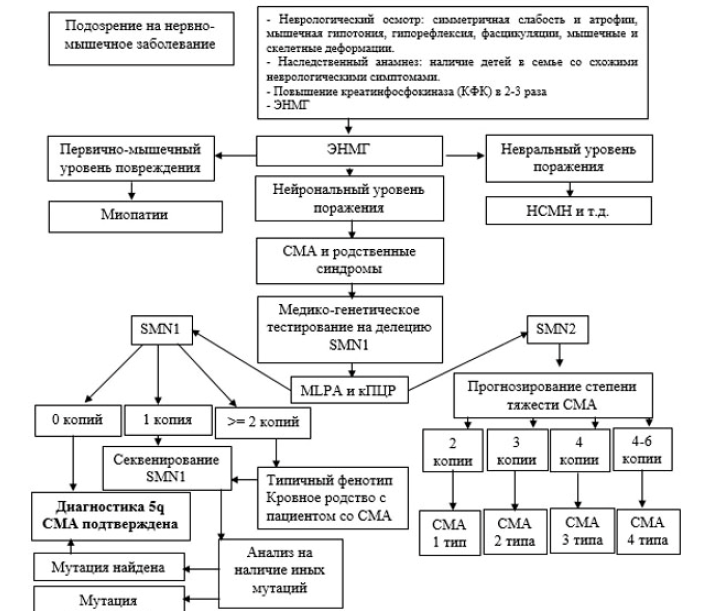
13.4.1. при подозрении на СМА 5q молекулярно-генетическое исследование генов SMN1/SMN2 включает определение числа копий экзонов 7, 8 гена SMN1, поиск точечных мутаций в гене SMN1 (при необходимости). При биаллельной (гомозиготной) делеции 7 или 7 и 8 экзонов гена SMN1 диагноз СМА подтвержден. Медицинские показания для проведения поиска точечных мутаций определяются врачебным консилиумом, если:
Диагноз СМА подтвержден при наличии:
Определение числа копий гена SMN2 и поиск делеций 7 и 8 экзонов в гене SMN2 проводится пациентам с подтвержденным диагнозом СМА с целью определения тактики лечения.
13.4.2. при подозрении на бульбоспинальную мышечную атрофию Кеннеди определяется число повторов в первом экзоне гена AR;
13.4.3. при подозрении на НМСН 1А – выявление дупликации или делеции в гене PMP22, расположенном на коротком плече 17 хромосомы. Дупликация гена PMP22 подтверждает диагноз НМСН 1А типа. Отсутствие изменений гена PMP22 не исключает диагноза НМСН 1А типа, поскольку данное заболевание является генетически гетерогенным и патогенная аберрация может быть обнаружена при дальнейшем исследовании.
13.4.4. при подозрении на МД Дюшенна-Беккера проводится поиск крупных делеций или дупликаций в гене DMD. При отсутствии в гене DMD крупных патогенных перестроек выполняется поиск точечных мутаций в гене DMD;
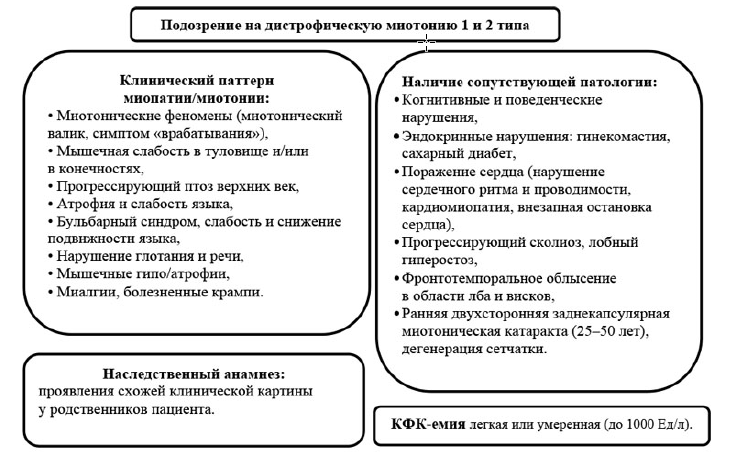
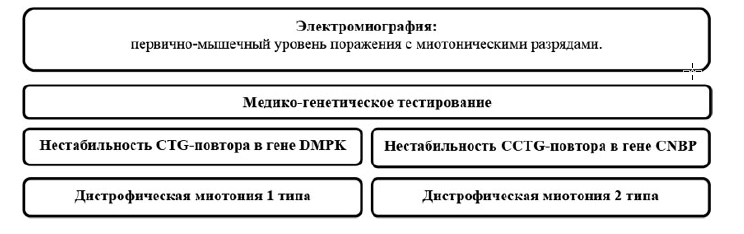
13.4.5. при подозрении на дистрофическую миотонию определяется количество CTG повторов в 3’-некодируемой области гена DMPK и CCTG повторов в интроне 1 гена CNBP (ZNF9).
13.4.6. при подозрении на окулофарингеальную мышечную дистрофию определяется число GCG повторов в первом экзоне гена PABPN1;
13.4.7. при подозрении на болезнь Помпе определяется активность кислой α-глюкозидазы в лимфоцитах крови. При положительном результате ферментного теста проводится поиск мутаций в гене GAA;
13.4.8. при невозможности идентифицировать патогенный вариант(ы), который(ые) с учетом клинических проявлений у пациента можно рассматривать как причину заболевания в качестве заключительного этапа диагностического поиска, рассматривают поиск точечных мутаций в генах, ассоциированных с ННМЗ, с использованием технологии высокопроизводительного секвенирования в масштабе таргетной панели, клинического или полного экзома, или генома. Решение о необходимости проведения высокопроизводительного секвенирования принимается врачебным консилиумом с участием главного внештатного генетика Министерства здравоохранения и профильного врача-специалиста РНПЦ неврологии и нейрохирургии при наличии медицинских показаний. Если применение всего комплекса диагностических молекулярно-генетических исследований диагноз ННМЗ не подтвердило, проводится повторное исследование фенотипа пациента и с интервалом 1 год выполняется повторный анализ данных секвенирования полного экзома (генома).
магнитно-резонансная томография мышц (по медицинским показаниям);
13.4. молекулярно-генетические диагностические исследования (медицинские показания и объем молекулярно-генетических исследований определяется врачом-генетиком):
13.4.1. при подозрении на СМА 5q молекулярно-генетическое исследование генов SMN1/SMN2 включает определение числа копий экзонов 7, 8 гена SMN1, поиск точечных мутаций в гене SMN1 (при необходимости). При биаллельной (гомозиготной) делеции 7 или 7 и 8 экзонов гена SMN1 диагноз СМА подтвержден. Медицинские показания для проведения поиска точечных мутаций определяются врачебным консилиумом, если:
- присутствует делеция 7 или 7 и 8 экзонов гена SMN1 в гетерозиготном состоянии при клиническом фенотипе, соответствующем СМА;
- отсутствует делеция 7 или 7 и 8 экзонов гена SMN1 у пациента с выраженным типичным фенотипом, характерным для СМА, и установленным кровным родством родителей.
Диагноз СМА подтвержден при наличии:
- делеции 7 или 7 и 8 экзонов гена SMN1 в гетерозиготном состоянии и патогенной точковой мутаций в транс положении;
- биаллельного(ых) патогенной(ых) точковой(ых) мутаций.
Определение числа копий гена SMN2 и поиск делеций 7 и 8 экзонов в гене SMN2 проводится пациентам с подтвержденным диагнозом СМА с целью определения тактики лечения.
Алгоритм диагностики пациентов со СМА и родственными синдромами установлен согласно приложению 1;
13.4.2. при подозрении на бульбоспинальную мышечную атрофию Кеннеди определяется число повторов в первом экзоне гена AR;
13.4.3. при подозрении на НМСН 1А – выявление дупликации или делеции в гене PMP22, расположенном на коротком плече 17 хромосомы. Дупликация гена PMP22 подтверждает диагноз НМСН 1А типа. Отсутствие изменений гена PMP22 не исключает диагноза НМСН 1А типа, поскольку данное заболевание является генетически гетерогенным и патогенная аберрация может быть обнаружена при дальнейшем исследовании.
Алгоритм диагностики НМСН установлен согласно приложению 2;
13.4.4. при подозрении на МД Дюшенна-Беккера проводится поиск крупных делеций или дупликаций в гене DMD. При отсутствии в гене DMD крупных патогенных перестроек выполняется поиск точечных мутаций в гене DMD;
13.4.5. при подозрении на дистрофическую миотонию определяется количество CTG повторов в 3’-некодируемой области гена DMPK и CCTG повторов в интроне 1 гена CNBP (ZNF9).
Алгоритм диагностики дистрофической миотонии установлен согласно приложению 3;
13.4.6. при подозрении на окулофарингеальную мышечную дистрофию определяется число GCG повторов в первом экзоне гена PABPN1;
13.4.7. при подозрении на болезнь Помпе определяется активность кислой α-глюкозидазы в лимфоцитах крови. При положительном результате ферментного теста проводится поиск мутаций в гене GAA;
13.4.8. при невозможности идентифицировать патогенный вариант(ы), который(ые) с учетом клинических проявлений у пациента можно рассматривать как причину заболевания в качестве заключительного этапа диагностического поиска, рассматривают поиск точечных мутаций в генах, ассоциированных с ННМЗ, с использованием технологии высокопроизводительного секвенирования в масштабе таргетной панели, клинического или полного экзома, или генома. Решение о необходимости проведения высокопроизводительного секвенирования принимается врачебным консилиумом с участием главного внештатного генетика Министерства здравоохранения и профильного врача-специалиста РНПЦ неврологии и нейрохирургии при наличии медицинских показаний. Если применение всего комплекса диагностических молекулярно-генетических исследований диагноз ННМЗ не подтвердило, проводится повторное исследование фенотипа пациента и с интервалом 1 год выполняется повторный анализ данных секвенирования полного экзома (генома).
Лечение
ГЛАВА 4
ЛЕЧЕНИЕ ННМЗ
14. Терапия ННМЗ подразделяется на два направления: патогенетическая и симптоматическая терапия (коррекция отдельных симптомов заболевания).
15. Патогенетическая терапия болезни Помпе: проведение ферментной заместительной терапии (далее – ФЗТ) с целью замедления прогрессирования болезни, улучшения состояния костно-мышечной и дыхательной системы, повышения выживаемости, удлинения периода жизни до наступления необходимости в вентиляции легких и кресле-коляске.
Назначается алглюкозидаза альфа, лиофилизат для приготовления концентрата для приготовления раствора для инфузий 50 мг, вводится внутривенно капельно 20 мг/кг 1 раз в 2 недели. Первые 3 инфузии проводятся в больничной организации с мониторингом витальных функций (частота сердечных сокращений, артериальное давление, сатурация) во время инфузии. В дальнейшем ФЗТ назначается по медицинским показаниям при осложненном течении болезни, проводится в условиях круглосуточного пребывания в больничной организации, при стабильном состоянии пациента – в организациях здравоохранения дневного пребывания или амбулаторно-поликлинических организациях 1 раз в 2 недели.
16. Прививки при ННМЗ не противопоказаны. Вакцинация проводится по индивидуальному графику, включающему наряду с мероприятиями Национального календаря профилактических прививок сезонную вакцину от гриппа и пневмококковую вакцину.
17. При симптоматической терапии ННМЗ назначаются следующие ЛП:
17.1. один из нейропротекторов:
- этилметилгидроксипиридина сукцинат, таблетки, покрытые оболочкой 125 мг, таблетки, покрытые оболочкой 250 мг, внутрь 250–500 мг в сутки в 2–3 приема;
- максимальная суточная доза – 600–800 мг; раствор для инъекций 50 мг/мл 2, 5 и 10 мл, при внутримышечно или внутривенно введении разовая доза 50–400 мг, максимальная суточная доза – 1200 мг;
- метилэтилпиридинола гидрохлорид, раствор для инъекций 10 мг/мл, 5 мл внутримышечно или 10 мл внутривенно;
- глицин, таблетки подъязычные 100 мг, сублингвально или трансбуккально по 100 мг 3 раза в сутки;
17.2. один из парасимпатомиметических ЛП:
- холина альфосцерат, капсулы 400 мг, 600 мг, порошок для приготовления раствора для приема внутрь 400 мг, 600 мг, по 400 мг 3 раза в сутки или 600 мг 2 раза в сутки; раствор для инъекций 250 мг/1 мл – 4 мл (1000 мг) внутримышечно или внутривенно;
- ипидакрин, таблетки 20 мг, по 0,5–1 таблетке 1–3 раза в сутки; раствор для инъекций 1,5 %–1,0, при внутримышечном введении разовая доза 5–30 мг, максимальная доза 200 мг в сутки;
17.3. один из метаболических ЛП:
- мельдоний, капсулы 250 мг, капсулы 500 мг, внутрь по 500 мг – 1 г/сут, раствор для инъекций 10 % 5 мл внутривенно или внутримышечно;
- тиоктовая кислота, таблетки, покрытые оболочкой 600 мг, внутрь по 600 мг 1 раз в сутки, раствор для внутривенно инъекций 25 мг/мл – 24 мл, 600 мг в сутки;
17.4. один из периферических вазодилататоров:
- пентоксифиллин, таблетки, покрытые оболочкой 100 мг, 200 мг, внутрь 3 раза в сутки; таблетки, покрытые оболочкой ретард 400 мг, 600 мг, внутрь 2 раза в сутки;
- ксантинола никотинат, таблетки 150 мг, внутрь 3 раза в сутки; раствор для инъекций 15 % – 2,0, внутримышечно 1–3 раза в сутки;
- ницерголин, капсулы 10 мг, 30 мг, таблетки, покрытые оболочкой 5 мг, 10 мг, 30 мг, внутрь по 5–10 мг 3 раза в сутки;
17.5. при умеренном и выраженном псевдобульбарном синдроме назначается один из следующих ЛП:
- флуоксетин, капсулы 20 мг, начальная доза 20 мг в сутки, при необходимости доза может быть увеличена до 40–60 мг в сутки в 1–2 приема; максимальная суточная доза 80 мг в 1–2 приема;
- амитриптилин, таблетки 25 мг, на ночь внутрь в дозе 12,5 мг. Эффективная доза подбирается методом титрования, увеличивая дозу на 12,5 мг раз в 1–3 дня. Суточная доза 12,5–50 мг;
- лоразепам, таблетки, покрытые оболочкой, 1 мг, 2,5 мг, внутрь 0,5–1,25 мг 2 раза в сутки;
- пароксетин, таблетки, покрытые пленочной оболочкой, 20 мг, 30 мг внутрь, стартовая доза 5–10 мг один раз в сутки в течение недели, далее увеличение дозы на 10 мг до 20 мг в сутки; максимальная суточная доза 60 мг;
17.6. противоэпилептические ЛП:
- карбамазепин, таблетки, 200 мг, стартовая доза 100 мг внутрь 2 раза в сутки.
Эффективная доза подбирается методом титрования, увеличивая суточную дозу не более чем на 200 мг до достижения эффекта; суточная доза составляет 600–1200 мг в 2–3 приема;
17.7. при гиперсаливации применяется один из следующих ЛП:
- амитриптилин, таблетки 25 мг, на ночь внутрь в дозе 12,5 мг. Эффективная доза подбирается методом титрования, увеличивая дозу на 12,5 мг раз в 1–3 дня; суточная доза 12,5–50 мг;
- ботулинический токсин типа А, инъекции в слюнные железы с целью уменьшения выраженности этого симптома под контролем УЗИ слюнных желез;
17.8. при вязкой мокроте назначается ацетилцистеин (таблетки шипучие, 200 мг, 200 мг) 3 раза в сутки (не позднее 6 часов до сна) внутрь или 600 мг 1 раз в сутки утром;
17.9. при болезненных мышечных спазмах (крампи) и (или) фасцикуляциях назначается один из следующих ЛП:
- карбамазепин (таблетки, 200 мг), стартовая доза 100 мг внутрь 2 раза в сутки. Эффективная доза подбирается методом титрования, увеличивая суточную дозу не более чем на 200 мг до достижения эффекта; суточная доза составляет 600 мг – 1200 мг в 2–3 приема;
- окскарбазепин (таблетки 150 мг, 300 мг и 600 мг). Лечение начинается с дозы 300 мг 1–2 раза в сутки. Дальнейшее увеличение на 300 мг на каждый прием 1 раз в неделю. Терапевтическая доза 600–2400 мг в сутки;
- ламотриджин (таблетки (таблетки жевательные/растворимые) 25 мг; таблетки 50 мг; таблетки (таблетки жевательные/растворимые) 100 мг). Лечение начинается с дозы 25 мг 1 раз в сутки. Дальнейшее увеличение на 25 мг в сутки 1 раз в 2 недели до 50 мг в сутки. Дальнейшее увеличение возможно в дозе 50 мг в сутки. Терапевтическая доза 200 мг в сутки за 2 приема;
17.10. при спастическом синдроме назначается баклофен (таблетки 10 мг, таблетки 25 мг), стартовая доза 5 мг 3 раза в сутки, через три дня приема разовая доза увеличивается вдвое, в последующие три дня разовая доза увеличивается до 15 мг, в течение следующих 3 дней – до 20 мг, в случае клинической необходимости суточная доза может быть с осторожностью увеличена до 100 мг;
17.11. при нейропатическом болевом синдроме назначается один из следующих ЛП:
- карбамазепин (таблетки, 200 мг), стартовая доза 100 мг внутрь 2 раза в сутки. Эффективная доза подбирается методом титрования, увеличивая суточную дозу не более чем на 200 мг до достижения эффекта; суточная доза составляет 600 мг – 1200 мг;
- габапентин (капсулы 300 мг), в дозе 300 мг внутрь, начиная с вечернего приема. Эффективная доза подбирается методом титрования, увеличивая суточную дозу на 300 мг каждые 2–3дня до достижения эффекта. Эффективная суточная доза составляет 1200–1800 мг; Максимальная суточная доза составляет 3600 мг;
- прегабалин (капсулы 75 мг, капсулы 150 мг), в дозе 75 мг внутрь 1–2 раза в день. Эффективная доза подбирается методом титрования, через 3–7 дней доза может быть увеличена до 300 мг в сутки в 2–3 приема, в случае необходимости через 7 дней может быть увеличена до максимальной дозы 600 мг в сутки в 2–3 приема;
- амитриптилин (таблетки 25 мг), на ночь внутрь в дозе 12,5 мг. Эффективная доза подбирается методом титрования, увеличивая дозу на 12,5 мг раз в 1–3 дня; Суточная доза 12,5–50 мг;
- венлафаксин (таблетки и капсулы 37,5 мг, 75 мг, 150 мг). Начальная доза составляет 37,5–75 мг в сутки, таблетки принимаются 2 раза в сутки, капсулы – 1 раз в сутки. Для достижения клинического эффекта дозу повышают на 75 мг один раз в 7–14 дней в 2 приема для таблеток, в 1 прием – для капсул. Противоболевой эффект лучше проявляется в дозе 225 мг в сутки. Максимальная суточная доза – 375 мг;
- дулоксетин (капсулы кишечнорастворимые 30 мг, капсулы кишечнорастворимые 60 мг), назначается в разовых дозах 30–60 мг внутрь, 1–2 раза в сутки; суточная доза составляет 60–120 мг;
17.12. при депрессивном, тревожном и астеническом расстройстве назначается:
17.12.1. один из антидепрессантов – селективных ингибиторов хобратного захвата серотонина (далее – СИОЗС):
- сертралин (таблетки, покрытые пленочной оболочкой, 25 мг, 50 мг, 100 мг внутрь), стартовая доза 25 мг внутрь один раз в сутки, через неделю дозу повышают до 50 мг в сутки в 1 прием, при недостаточном эффекте через 6 недель приема увеличить дозу на 25 мг за 1–2 недели до 75–100 мг в сутки, максимальная суточная доза 200 мг;
- пароксетин (таблетки, покрытые пленочной оболочкой, 20 мг, 30 мг), внутрь, стартовая доза 5–10 мг один раз в сутки в течение недели, далее – 20 мг в сутки в 1 прием, при недостаточном эффекте через 6 недель приема увеличить дозу на 10 мг за 1–2 недели до средней эффективной дозы 30–40 мг в сутки, максимальная суточная доза 60 мг;
- флувоксамин (таблетки, покрытые пленочной оболочкой, 50 мг, 100 мг), внутрь стартовая доза 25 мг один раз в сутки в течение недели, далее 50 мг в сутки на ночь, при недостаточном эффекте через 6 недель приема постепенно увеличить дозу на 25–50 мг за 1–2 недели до 100 мг в сутки (в два приема), максимальная суточная доза 200 мг;
- эсциталопрам (таблетки, покрытые пленочной оболочкой, 10 мг), внутрь, стартовая доза 2,5–5 мг один раз в сутки в течение недели, далее 10 мг в сутки, при недостаточном эффекте через 6 недель приема увеличение дозы на 5–10 мг за 1–2 недели, максимальная суточная доза 20 мг;
- флуоксетин (капсулы 20 мг), начальная доза 20 мг в сутки, при необходимости доза может быть увеличена до 40–60 мг в сутки в 1–2 приема; максимальная суточная доза 80 мг в 1–2 приема.
Длительность лечения антидепрессантами зависит от клинического состояния и продолжается от 4–6 месяцев (контроль состояния не реже 1 раза в месяц), при необходимости – несколько лет или постоянно. Отмена осуществляется постепенно в течение 2–4 недель;
17.12.2. в целях предупреждения нежелательных лекарственных реакций, а также для купирования клинически выраженной тревоги в первые 1–2 недели рекомендуется назначать СИОЗС в комбинации с одним из бензодиазепинов:
- диазепам (таблетки (таблетки, покрытые оболочкой) 5 мг, раствор для внутривенного и внутримышечного введения (для инъекций) 5 мг/мл 2 мл), стартовая разовая доза 5 мг 1–3 раза в сутки, средняя терапевтическая доза 30 мг в сутки, максимальная суточная доза 60 мг;
- лоразепам (таблетки, покрытые оболочкой, 1 мг, таблетки, покрытые оболочкой, 2,5 мг), внутрь разовая доза 0,5–1,25 мг 1–3 раза в сутки. Средняя терапевтическая доза 5 мг в сутки. Максимальная суточная доза 10 мг;
- клоназепам (таблетки 0,5 мг, 2 мг), стартовая разовая доза 0,25–0,5 мг 2–4 раза в сутки, при недостаточном противотревожном эффекте постепенное увеличивают дозу на 0,25–1 мг раз в 3 дня до средней терапевтической доза 2–4 мг в сутки, максимальная суточная доза 8 мг;
17.12.3. при сочетании депрессии и (или) тревоги с невропатической болью используется один из антидепрессантов с дополнительным противоболевым эффектом:
- амитриптилин (таблетки 25 мг), на ночь внутрь в дозе 12,5 мг. Эффективная доза подбирается методом титрования, увеличивая дозу на 12,5 мг раз в 1–3 дня. Суточная доза 12,5–50 мг;
- венлафаксин (таблетки и капсулы 37,5 мг, 75 мг, 150 мг). Начальная доза составляет 37,5–75 мг в сутки, таблетки принимаются 2 раза в сутки, капсулы – 1 раз в сутки. Для достижения клинического эффекта дозу повышают на 75 мг один раз в 7–14 дней в 2 приема для таблеток, в 1 прием для капсул. Противоболевой эффект максимально проявляется в дозе 225 мг в сутки. Максимальная суточная доза – 375 мг;
- дулоксетин (капсулы кишечнорастворимые 30 мг, 60 мг), в разовых дозах 30–60 мг внутрь, 1–2 раза в сутки; суточная доза составляет 60–120 мг;
17.12.4. при нарушении сна на фоне депрессивного, тревожного и астенического расстройства применяется один из следующих ЛП:
- миртазапин (таблетки, покрытые пленочной оболочкой, 30 мг, 45 мг), внутрь стартовая доза 15 мг (предпочтительно один раз в сутки, перед сном), при необходимости увеличить дозу на 15 мг в неделю до 45 мг в сутки;
- мапротилин (таблетки, покрытые оболочкой 25 мг), внутрь стартовая доза 12,5–25 мг один раз в сутки, при недостаточном эффекте увеличение дозы на 12,5–25 мг один раз в 5–7 дней. Средняя терапевтическая доза 75 мг в сутки (кратность применения 1–3 раза в сутки), максимальная суточная доза 150 мг.
Если ожидаемая продолжительность жизни пациента меньше 4 недель используется монотерапия бензодиазепинами;
17.12.5. при неэффективности бензодиазепинов и антидепрессантов терапию дополняют одним из антипсихотических ЛП:
- оланзапин (таблетки 5 мг, 10 мг), внутрь стартовая доза 2,5 мг один раз в сутки (вечером), при необходимости постепенное увеличение дозы на 2,5 мг до 10 мг в сутки;
- кветиапин (таблетки, покрытые пленочной оболочкой, 25 мг, 200 мг), внутрь стартовая доза 12,5 мг однократно на ночь, далее ежедневное увеличение суточной дозы на 12,5 мг до достижения эффективной дозы, средняя терапевтическая доза 200 мг в сутки, максимальная суточная доза 400 мг;
- хлорпротиксен (таблетки, покрытые пленочной оболочкой 15 мг, 25 мг, 50 мг), внутрь стартовая доза 15 мг один раз в сутки (вечером), при необходимости постепенное увеличение дозы, средняя терапевтическая доза 75 мг в сутки, максимальная суточная доза 150 мг.
18. ЛП (пищевые добавки), обеспечивающие минерализацию костей:
- холекальциферол (таблетки, 500 МЕ), профилактическая доза внутрь от 500 ME до 1000 ME в сутки; начальную терапию дефицита витамина D проводят в дозе 4000 ME в сутки в течение 6–12 недель, а затем переходят к поддерживающей терапии в дозах 1500–2000 ME в сутки или кальция глюконат (таблетки 500 мг), по одной таблетке 1–3 раза в сутки.
19. Терапия глюкокортикоидами системного действия МД Дюшенна и МД Беккера: продолжение приема преднизолона 0,75 мг/кг/сутки внутрь по достижении 18-летнего возраста по схеме, назначенной в детском возрасте для стабилизации функционального состояния пациентов.
20. Основные принципы очищения дыхательных путей: сочетанное применение механического инсуфлятора-аспиратора (откашливателя), ручных компрессий грудной клетки, постурального дренажа с электрическим аспиратором для дренирования мокроты у лежачих и сидячих пациентов.
Относительное медицинское противопоказание к использованию инсуфлятора-аспиратора – эмфизема. Показателем эффективности проводимых мероприятий являются данные оксиметрии.
21. Неинвазивная вентиляция легких (далее – НИВЛ) с двухуровневым положительным давлением (BIPAP) показана всем пациентам с ННМЗ с симптомами дисфункции дыхательной мускулатуры для медицинской профилактики гипотрофии грудной клетки, облегчения одышки и борьбы с гиповентиляцией.
22. Медицинские показания для начала НИВЛ:
22.1. клинические признаки дыхательной недостаточности:
- жалобы на диспноэ, ортопноэ, тахипноэ;
- нарушения сна вследствие ночной одышки;
- утренние головные боли;
- участие в акте дыхания вспомогательной дыхательной мускулатуры в покое;
- наличие парадоксального дыхания;
- чрезмерная дневная сонливость;
22.2. лабораторные показатели дыхательной недостаточности:
- жизненная емкость легких (далее – ЖЕЛ) <80 % должного объема при наличии клинических симптомов;
- ЖЕЛ <50 % должного объема без клинических симптомов или ее быстрое снижение;
- РаО2 <60 мм рт. ст.;
- десатурация в ночное время выраженной степени, более 5 эпизодов десатурации в течение часа по данным ночной оксиметрии (ODI >5/ч);
- хроническая дневная гиперкапния с PaCO 2 >45 мм рт. ст.; ночная гиперкапния с PaCO 2 >50 мм рт. ст.; дневная нормокапния с ростом PaCO2 ночью на 10 мм рт. ст. и более.
23. Медицинские противопоказания для НИВЛ:
- выраженная секреция слизи в дыхательных путях, хроническая аспирация;
- невозможность защитить дыхательные пути;
- непродуктивный кашель;
- нарушение сознания, кома, изменения ментального статуса;
- острая травма или анатомические нарушения лица;
- необходимость постоянной вентиляционной поддержки;
- развитие критического состояния.
НИВЛ рекомендовано сочетать с техникой очищения дыхательных путей пациентам при сохранной функции ходьбы ходячим и сидячим пациентам с ННМЗ в острых ситуациях (инфекция дыхательных путей) с целью медицинской профилактики развития осложнений. Применение ингаляции чистого кислорода без дополнительной вентиляции легких не показано (может привести к нарастанию респираторного ацидоза).
24. Искусственная вентиляция легких при ННМЗ проводится при неэффективности НИВЛ для коррекции дыхательных нарушений (постановка временной или постоянной трахеостомы).
25. Ортопедическая медицинская помощь пациентам с ННМЗ:
- консультация врача-травматолога-ортопеда 1 раз в 6 месяцев;
- использование жестких ортезов-туторов на нижние конечности с шарнирами на уровне тазобедренных, коленных и голеностопных суставов для нестоящих пациентов;
- использование жестких ортезов-корсетов на позвоночник для сидячих и ходящих пациентов;
- использование специальной обуви, ортезов голеностопного сустава/стопы для исправления свисания стопы и облегчения ходьбы;
- выполнение заднего спондилодеза, хирургическое лечение нестабильности тазобедренного сустава, контрактур суставов и деформации конечностей; при переломах длинных (трубчатых) костей нижних конечностей для ходячих пациентов и с переломами бедра для лежачих пациентов экстракортикальный остеосинтез, интрамедуллярный блокируемый остеосинтез, интрамедуллярный спицевой/стержневой остеосинтез.
26. Гастроэнтерологическая (диетологическая) медицинская помощь пациентам с ННМЗ:
26.1. проведение оценки функции желудочно-кишечного тракта (далее – ЖКТ) с участием врача-гастроэнтеролога и (или) врача-диетолога: контроль массы тела, оценка состояния функции глотания;
26.2. определение типа рациона и способа питания с учетом индивидуальной переносимости:
- обеспечение питания пациента с интервалом 6 часов с включением в рацион источника белка во избежание голодания и для предотвращения метаболического ацидоза, нарушений обмена жирных кислот, гипер- и гипогликемии;
- восполнение потери жидкости;
- прием прокинетиков и пробиотиков для улучшения моторики ЖКТ;
- прием пищи полутвердой консистенции при дисфагии для предотвращения аспирации;
- использование желеобразных форм, жидкие пюре, загустители жидкости на основе мальтодекстрина промышленного производства;
- использование смеси для энтерального питания при кормлении через зонд или гастростому.
27. Тактика ведения пациентов с дисфагией, утративших способность сидеть:
- анализ режима питания;
- антропометрия 1 раз в 6 месяцев;
- исследование уровня 25-гидроксивитамина D;
- денситометрия;
- оценка моторной функции ЖКТ;
- по медицинским показаниям рекомендовать прием ЛП для коррекции выявленных нарушений;
- установка в превентивных целях назоеюнального зонда до возможности установки желудочного зонда с фундопликацией и гастростомией при дефиците индекса массы тела;
- проведение коррекции калорийности и количества потребляемой жидкости, макро-и микроэлементов, уровня глюкозы, уровня потребления кальция и витамина D с участием врача-гастроэнтеролога, врача-диетолога.
28. Тактика ведения пациентов с дисфагией при сохранении способности сидеть:
- обеспечение в активной стадии заболевания оптимального потребления жидкости, кальция и витамина D, контроль уровня глюкозы, включение в рацион большого количества клетчатки;
- установка носового зонда до возможности установки долгосрочного желудочного зонда, если пациент не способен самостоятельно глотать;
- проведение совместно с врачом-гастроэнтерологом, врачом-диетологом коррекции калорийности потребляемой пищи, количества потребляемой жидкости, макро-и микроэлементов при дефиците массы тела, ограничение калорийности для пациентов с избыточной массой тела.
29. Тактика ведения пациентов с дисфагией при сохранной функции ходьбы:
- проведение анализа питания при недостаточной или избыточной массе тела;
- ежегодная антропометрия (рост, масса тела);
- контроль метаболизма глюкозы, уровня 25-гидроксивитамина D, макро-и микроэлементов;
- установка назогастрального зонда (до 4 недель) с последующим обеспечением постоянного кормления через гастростому;
- обучение пациента и ухаживающих лиц правилам ухода и кормления в домашних условиях.
31. Медицинские противопоказания для чрескожной эндоскопической гастростомии:
30. Медицинские показания для применения чрескожной эндоскопической гастростомии:
- дисфагия и невозможность адекватного и безопасного кормления через рот;
- потеря веса >10 % в связи с нарушенным глотанием;
- приближение величины ЖЕЛ к 50 %.
31. Медицинские противопоказания для чрескожной эндоскопической гастростомии:
- стеноз глотки, пищевода;
- выраженное ожирение, асцит, портальная гипертензия, язвенная болезнь желудка, геморрагический диатез, выраженная слабость диафрагмы;
- выраженные дыхательные нарушения (относительное медицинское противопоказание ЖЕЛ <30 %, при ЖЕЛ <15 % установка может быть уже нецелесообразной);
- сепсис, перитонит;
- общее тяжелое состояние пациента;
- оперированный желудок;
- соматическая, инфекционная патология в стадии обострения;
- невозможность проведения эндоскопа в желудок.
Медицинская реабилитация
ГЛАВА 5
МЕДИЦИНСКАЯ РЕАБИЛИТАЦИЯ ПАЦИЕНТОВ С ННМЗ И ОКАЗАНИЕ ПАЛЛИАТИВНОЙ МЕДИЦИНСКОЙ ПОМОЩИ
32. Реабилитационные мероприятия и обучение пациентов с ННМЗ проводятся в зависимости от стадии заболевания и ведущих клинических симптомов.
33. Физическая реабилитация проводится на ранних стадиях заболевания в следующих целях:
- профилактика контрактур, мышечно-скелетной боли;
- поддержание силы мышц и объема движений;
- оптимизация техники ходьбы, тренировка ходьбы со вспомогательными техническими средствами (тростями, ходунками, ортезами);
- обучение техникам сохранения энергии;
- продление периода самостоятельности пациента в повседневной жизни.
34. Методы физической реабилитации:
- лечебная физическая культура (далее – ЛФК);
- длительное вытяжение с использованием позиционирования, шинирования;
- гидрокинезиотерапия;
- механотерапия, безнагрузочная механотерапия, роботизированная механотерапия, аппаратные стато-кинетические нагрузки, тренировки с биологической обратной связью по координации движений (стабилоплатформа), имитация ходьбы со стабилизацией;
- вертикализация с использованием приспособлений (вертикализатор, кровать-вертикализатор, параподиум-вертикализатор), ходьбы и ортопедическая обувь;
- ортезирование и тейпирование.
35. Рекомендуемые группы упражнений при ННМЗ:
- динамические аэробные упражнения с акцентом на аксиальную мускулатуру, тазовый пояс, бедра и голени;
- тренировка баланса сидя и стоя;
- упражнения для амортизации стоп;
- укрепление стабилизаторов туловища;
- коррекция статодинамического стереотипа (навык правильной позы сидя и стоя);
- дыхательные упражнения;
- упражнения на растягивание с сопротивлением низкой интенсивности с учетом силы мышц.
36. Проведение ЛФК включает следующие аспекты:
- умеренная интенсивность физических нагрузок;
- ограничение времени выполнения одного упражнения (на одну группу мышц);
- плавность и низкий темп движений во избежание провоцирования мышечных спазмов;
- индивидуальный подход к каждому пациенту.
37. Цели физической реабилитации на поздних стадиях заболевания:
- обучение пациента и членов семьи техникам правильного позиционирования и безопасного перемещения;
- использование вспомогательных технических средств (для надевания одежды, поручней, насадки на унитаз, сиденья для ванной, кресла-туалета и иное);
- при нарастании у пациента мышечной слабости – использование механического кресла-коляски.
38. Эрготерапевтическая коррекция при ННМЗ:
- оценка независимости в повседневной жизни (индекс Бартел) каждые 3 месяца;
- оценка домашней обстановки каждые 6 месяцев;
- рекомендации и обучение по использованию вспомогательных техник и приспособлений с целью облегчения повседневной активности.
39. Логопедическая коррекция при ННМЗ:
- оценка речевой функции каждые 3–6 месяцев;
- занятия с логопедом;
- оценка когнитивных способностей при нарушении речевой функции на ранних стадиях заболевания;
- использование вспомогательных приспособлений (таблицы, схемы) и приборов (специальные программы на телефонах и компьютерах) с целью облегчения коммуникации.
40. Оказание паллиативной медицинской помощи пациентам с ННМЗ осуществляется в порядке, определенном Министерством здравоохранения.
Информация
Источники и литература
-
Постановления и приказы Министерства здравоохранения Республики Беларусь об утверждении клинических протоколов 2025
-
www.minzdrav.gov.by
Информация
ПОСТАНОВЛЕНИЕ МИНИСТЕРСТВА ЗДРАВООХРАНЕНИЯ
РЕСПУБЛИКИ БЕЛАРУСЬ
13 мая 2025 г. № 42
Об утверждении клинического протокола
На основании абзаца девятого части первой статьи 1 Закона Республики Беларусь от 18 июня 1993 г. № 2435-XII «О здравоохранении», подпункта 8.3 пункта 8 и подпункта 9.1 пункта 9 Положения о Министерстве здравоохранения Республики Беларусь, утвержденного постановлением Совета Министров Республики Беларусь от 28 октября 2011 г. № 1446, Министерство здравоохранения Республики Беларусь
ПОСТАНОВЛЯЕТ:
1. Утвердить клинический протокол «Диагностика и лечение пациентов с наследственными нервно-мышечными заболеваниями (взрослое население)» (прилагается).
2. Настоящее постановление вступает в силу после его официального опубликования.
Министр А.В.Ходжаев
СОГЛАСОВАНО
Брестский областной исполнительный комитет
Витебский областной исполнительный комитет
Гомельский областной исполнительный комитет
Гродненский областной исполнительный комитет
Минский областной исполнительный комитет
Минский городской исполнительный комитет
Могилевский областной исполнительный комитет
Государственный пограничный комитет Республики Беларусь
Комитет государственной безопасности Республики Беларусь
Министерство внутренних дел Республики Беларусь
Министерство обороны Республики Беларусь
Министерство по чрезвычайным ситуациям Республики Беларусь
Национальная академия наук Беларуси
Управление делами Президента Республики Беларусь
Алгоритм диагностики пациентов со СМА и родственными синдромами
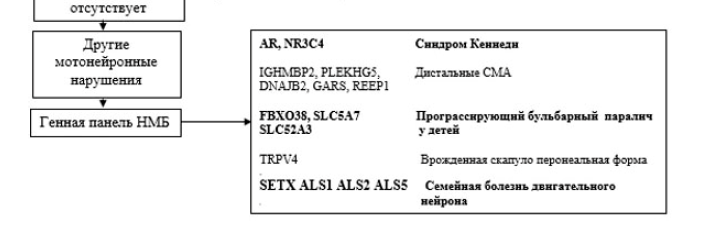
Алгоритм диагностики НМСН
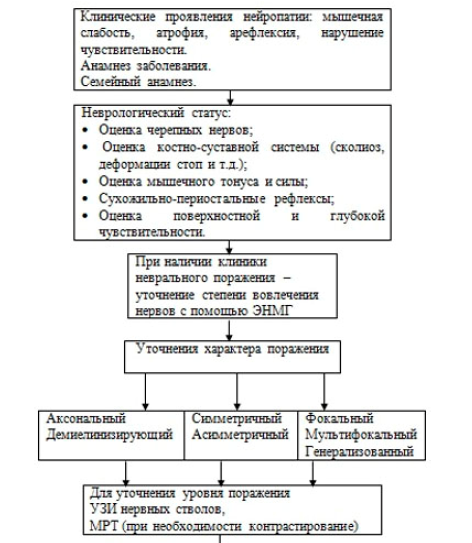
Алгоритм диагностики дистрофической миотонии
Приложение 1
к клиническому протоколу «Диагностика и лечение пациентов с наследственными нервно-мышечными заболеваниями (взрослое население)»
Алгоритм диагностики пациентов со СМА и родственными синдромами
Приложение 2
к клиническому протоколу «Диагностика и лечение пациентов с наследственными нервно-мышечными заболеваниями (взрослое население)»
Алгоритм диагностики НМСН
Приложение 3
к клиническому протоколу «Диагностика и лечение пациентов с наследственными нервно-мышечными заболеваниями (взрослое население)»
Алгоритм диагностики дистрофической миотонии
Прикреплённые файлы
Внимание!
- Занимаясь самолечением, вы можете нанести непоправимый вред своему здоровью.
- Информация, размещенная на сайте MedElement и в мобильных приложениях "MedElement (МедЭлемент)", "Lekar Pro", "Dariger Pro", "Заболевания: справочник терапевта", не может и не должна заменять очную консультацию врача. Обязательно обращайтесь в медицинские учреждения при наличии каких-либо заболеваний или беспокоящих вас симптомов.
- Выбор лекарственных средств и их дозировки, должен быть оговорен со специалистом. Только врач может назначить нужное лекарство и его дозировку с учетом заболевания и состояния организма больного.
- Сайт MedElement и мобильные приложения "MedElement (МедЭлемент)", "Lekar Pro", "Dariger Pro", "Заболевания: справочник терапевта" являются исключительно информационно-справочными ресурсами. Информация, размещенная на данном сайте, не должна использоваться для самовольного изменения предписаний врача.
- Редакция MedElement не несет ответственности за какой-либо ущерб здоровью или материальный ущерб, возникший в результате использования данного сайта.