Гипераммониемия. Нарушение цикла мочевины
![]() Версия: Клинические протоколы МЗ РК - 2023 (Казахстан)
Версия: Клинические протоколы МЗ РК - 2023 (Казахстан)
Общая информация
Краткое описание
КЛИНИЧЕСКИЙ ПРОТОКОЛ ДИАГНОСТИКИ И ЛЕЧЕНИЯ
Коды МКБ-10:
|
Код
|
Название |
| Е72 | Другие нарушения обмена аминокислот |
| Е72.2 | Нарушения обмена цикла мочевины |
| Е72.4 | Нарушения обмена орнитина |
Категория пациентов: дети.
РКИ – Рандомизированное контролируемое исследование;
ТМС – Тандемная масс-спектрометрия;
ASS – Argininosuccinate synthetase deficiency (дефицит аргининосукцинатсинтетазы);
NAGS – N-acetyl glutamate synthetase deficiency (дефицит N-ацетилглутаматсинтетазы);
OTC – Ornithine transcarbamylase (OTC) deficiency (дефицит орнитинтранскарбамилазы).
CTLN1 – цитруллинемия I типа
Шкала уровня доказательности:
| А | Высококачественный мета-анализ, систематический обзор РКИ или крупное РКИ с очень низкой вероятностью (++) систематической ошибки результаты которых могут быть распространены на соответствующую популяцию. |
| В | Высококачественный (++) систематический обзор когортных или исследований случай-контроль или Высококачественное (++) когортное или исследований случай-контроль с очень низким риском систематической ошибки или РКИ с невысоким (+) риском систематической ошибки, результаты которых могут быть распространены на соответствующую популяцию. |
| С | Когортное или исследование случай-контроль или контролируемое исследование без рандомизации с невысоким риском систематической ошибки (+). Результаты которых могут быть распространены на соответствующую популяцию или РКИ с очень низким или невысоким риском систематической ошибки (++ или +), результаты которых не могут быть непосредственно распространены на соответствующую популяцию. |
| D | Описание серии случаев или неконтролируемое исследование, или мнение экспертов. |
Классификация
Классификация
Клиническая классификация гипераммониемии по этиологии [2-3]
Первичная – НЦМ
Дефицит карбамилфосфатсинтетазы I (CPSI)
Дефицит орнитинтранскарбамилазы (OTC)
Дефицит аргининосукцинатсинтетазы (ASS) (также известный как классическая цитруллинемия или цитруллинемия I типа (CTLN1)
Дефицит аргининосукцинатлиазы (ASL) (также известный как аргининосукцинатная ацидурия)
Дефицит N-ацетилглутаматсинтетазы (NAGS)
Дефицит аргиназы
Вторичная – заболевания, сопровождающихся повышенным образованием аммиака или недостаточной его утилизацией (заболевания печени, сосудистые мальформации, инфекции мочевых путей, вызванной бактериями, продуцирующими уреазу (Proteus mirabilis и некоторые штаммы Klebsiella species), прием лекарственных препаратов).
Классификация по течению заболевания [4-6].
Типичная картина: симптомы проявляются в первые 24–48 часов после рождения. У младенца появляются симптомы после начала кормления, потому что грудное молоко или детская смесь обеспечивают белковую нагрузку. Начальные признаки включают сонливость, неспособность поддерживать нормальную температуру тела и плохой аппетит, за которыми обычно следуют рвота, вялость и кома, что идентично таковому у младенцев с сепсисом.
Атипичная картина (пациенты с частичным дефицитом ферментов):
хроническая рвота, задержка развития, судорожные припадки, нарушения сна.
головная боль, анорексия, рвота, вялость, атаксия после повышенного потребления белка или в периоды катаболического стресса
лабораторные отклонения.
Диагностика
Диагностические критерии [8-12].
Жалобы:
Анамнез:
Физикальное обследование:
Основные лабораторные исследования [13-15]:
Таблица 1. Возрастные нормы содержания аммиака в крови [10]
|
Возрастные группы
|
Содержание аммиака в крови в норме, мкмоль/л |
| Недоношенные новорожденные | 50–150 |
| Доношенные новорожденные | 64–110 |
| Старше 1 месяца | 21–50 |
|
NB!
Необходимы условия исследования уровня аммиака в крови:
образцы крови необходимо брать только из интактной (неповрежденной) периферической вены;
исследование должно осуществляться в первые 2 ч после забора крови (оптимально в течение 30 минут после забора крови);
нельзя проводить исследование крови при наличии гемолиза
|
• Тандемная масс-спектрометрия (ТМС): подтверждение генетических заболеваний.
Основные инструментальные исследования:
• Нейросонография: гидроцефалия.
• МРТ головного мозга: гипоксически-ишемическая энцефалопатия.
Дополнительные инструментальные исследования: нет.
Показания для консультации специалистов [14-15]:
Диагностический алгоритм:
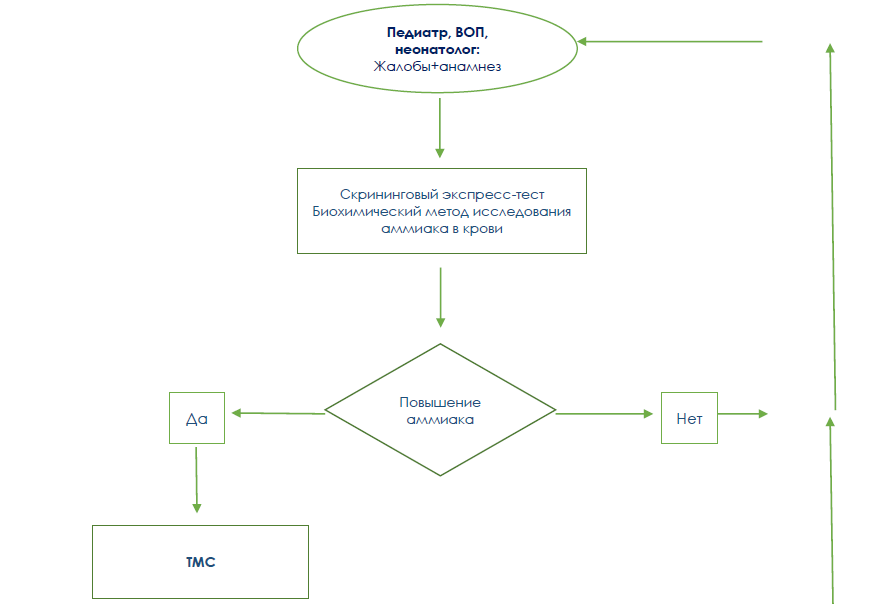
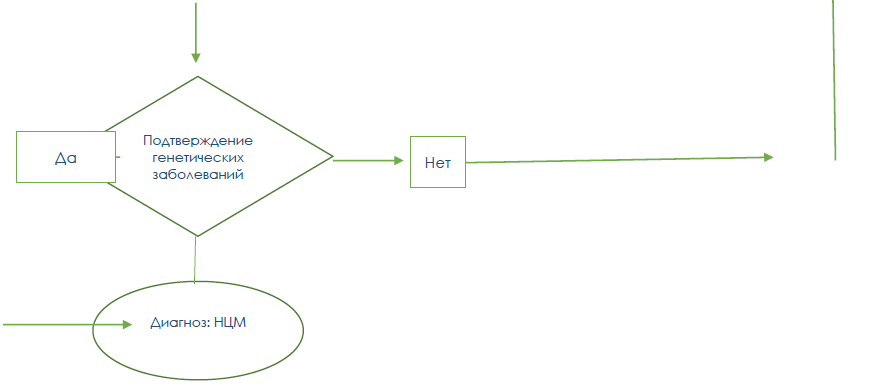
Дифференциальный диагноз
Дифференциальный диагноз и обоснование [16-17]:
|
Диагноз
|
Обоснование для дифференциальной диагностики | Обследования | Критерии исключения диагноза |
| Сепсис |
Схожая клиническая картина:
отказ от еды
нарушения ЦНС (летаргия, синдром угнетения/возбуждения)
респираторные нарушения
|
ОАК | не характерны воспалительные изменения в ОАК |
| С-реактивный белок | С-реактивный белок в пределах нормы | ||
| Прокальцитонин | прокальцитонин в пределах нормы | ||
| Бактериологический посев крови | отрицательное бактериологическое исследование крови | ||
| Рентгенография ОГК | Удовлетворительная пневматизация легких по рентгенографии | ||
| Гипоксически-ишемическая энцефалопатия тяжелой степени/кома | Схожая клиническая картина: нарушения ЦНС (летаргия, синдром угнетения/возбуждения) | Уточнение анамнеза | Отягощенный семейный анамнеза по наследственным заболеваниям. |
Лечение (амбулатория)
НЦМ – это группа генетических заболеваний, у которых нет этиологического лечения, терапия направлена на дезинтоксикацию (удаление аммиака из организма).
1. Немедикаментозное лечение [18-20]:
2. Медикаментозное лечение [18-20]:
Дезинтоксикационная терапия:
фенилбутират натрия: назначается с целью дезинтоксикации перорально, во время еды/кормления от 3 до 6 раз в день, терапия проводится пожизненно.
Перечень основных лекарственных средств:
|
Фармакотерапевтическая группа
|
МНН лекарственного средства | Способ применения | Уровень доказательности |
| Детоксикант аммония | Фенилбутират натрия* |
Младенцы и дети <20 кг: Перорально: от 450 до 600 мг/кг/день во время еды/кормления от 3 до 6 раз в день; максимальная суточная доза: 20 г/сут. Длительность курса: пожизнено.
Дети ≥20 кг: Перорально: от 9,9 до 13 г / м 2 /день в во время еды от 3 до 6 раз в день; максимальная суточная доза: 20 г / сут. Длительность курса: пожизнено. |
А[18-20] |
3. Хирургическое вмешательство: нет
4. Дальнейшее ведение [22]:
Профилактика последующих эпизодов гипераммониемии:
Пожизненный мониторинг: диспансерное наблюдение у педиатра, невролога.
Все пациенты, у которых диагностированы НЦМ должны быть обеспечены аммониеметрами.
Проведение лабораторного мониторинга показателей (измерение аммония плазмы в образцах венозной/капиллярной крови (целевой уровень <80 мкмоль/л):
при изменениях в диете, ухудшениях состояния ребенка – ежедневно;
Аммиак 2 раза в месяц в первые 2 месяца после постановки диагноза, далее 1 раз в 4 месяца и по показаниям.
Антропометрия (измерение роста и веса) – 1 раз в 4 месяца
|
NB!
Наибольший риск гипераммониемии в течение первого года жизни связан с вирусными заболеваниями (увеличивается катаболизм белков)!
|
избегать инфекционных контактов;
своевременная иммунизация в соответствии с Национальным календарем вакцинации РК;
В случаях, когда пероральный прием лекарств ограничен (из-за болезни, хирургического вмешательства) следует ввести внутривенное введение фенилацетата натрия-бензоата натрия и аргинина до тех пор, пока пероральная диета и лекарства не станут возможными. В эти периоды следует контролировать уровень аммиака.
Лабораторный мониторинг уровня аминокислот в крови.
Лечение (стационар)
ТАКТИКА ЛЕЧЕНИЯ НА СТАЦИОНАРНОМ УРОВНЕ
[21-26]
Подход к лечению заключается в следующем:
• прекращение потребления белка с целью сведения к минимуму катаболизма белка;
• регидратация;
• поддержание хорошего диуреза;
• дезинтоксикационная терапия.
1. Карта наблюдения пациента, маршрутизация пациента
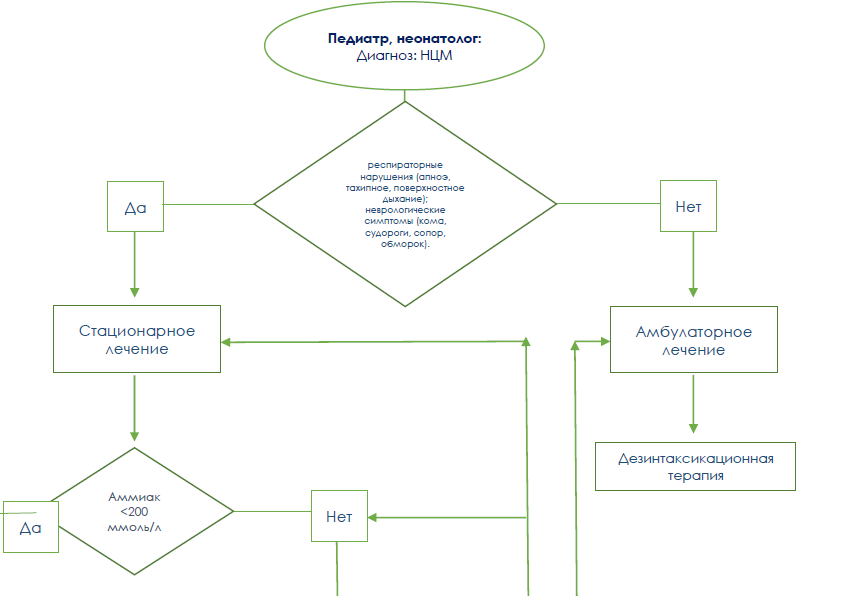
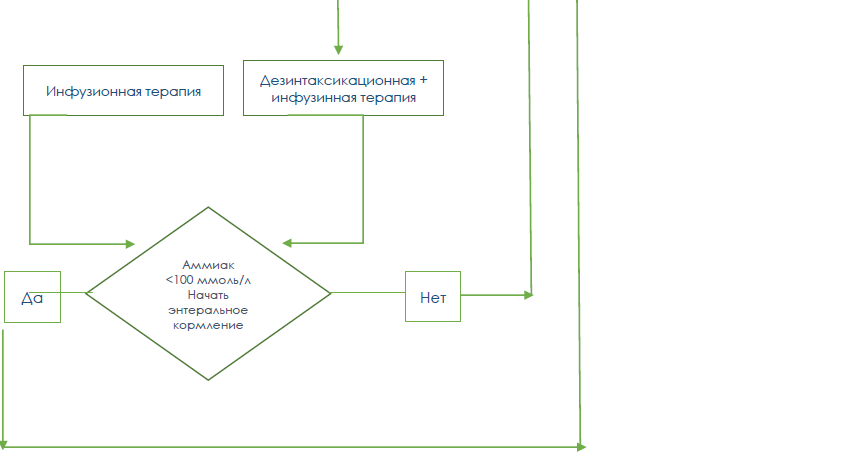
Энтеральное питание начинать при уровне аммиака <100 мкмоль/л. Назогастральный зонд может быть необходим для обеспечения надлежащего потребления. Младенцев кормят безбелковой смесью, в сочетании со смесями аминокислот и смесями на основе коровьего молока. Половину суточной потребности в белке обычно дают в виде смеси аминокислот, а половину — в виде белка коровьего молока. Рекомендуемое суточное потребление белка зависит от возраста и колеблется от 2–2,5 г/кг в день при рождении до менее 0,6–0,8 г/кг в день у детей старшего возраста.
при гипераммониемия <200 мкмоль/л – инфузионная терапия –;
при гипераммониемии >200 мкмоль/л – дезинтокикационная терапия + инфузионная терапия
Инфузионная терапия с целью обеспечения энергетической потребности и предотвращения развития катаболических процессов (необходимо обеспечить минимум 110% суточной потребности в энергии):
Дезинтоксикационная терапия проводится в два этапа:
1) внутривенная дезинтоксикационная терапия;
2) пероральная дезинтоксикационная терапия.
Внутривенная дезинтоксикационная терапия в зависимости от этиологии гипераммониемии:
• до тех пор, пока этиология заболевания не установлена: фенилацетата натрия-бензоата натрия и аргинина гидрохлорида;
• при NAGS, CPSI и OTC – аргинина гидрохлорида, фенилацетата натрия-бензоата натрия и цитруллин;
• при дефиците NAGS – карглуминовая;
• при дефиците ASS и ASL аргинина гидрохлорид и фенилацетат-натрия бензоат.
• при дефиците аргиназы – фенил ацетата.
при достижении уровня аммиака в крови менее 100 мкмоль/л;
переходе пациента на энтеральное питание
Перечень основных лекарственных средств [28-40]:
|
Фармакотерапевтическая группа
|
МНН лекарственного средства | Способ применения | Уровень доказательности |
| Детоксикант аммония | Фенилацетат натрия-бензоат натрия: * |
Младенцы и дети ≤20 кг:
Стартовая доза: внутривенно: 2,5 мл/кг (обеспечивает 250 мг/кг фенилацетата натрия и 250 мг/кг бензоата натрия) в течение 90–120 минут.
Поддерживающая доза:
Непрерывная внутривенная инфузия: 2,5 мл/кг в виде поддерживающей инфузии в течение 24 часов (обеспечивает 250 мг/кг фенилацетата натрия и 250 мг/кг бензоата натрия в течение 24 часов).
Дети и подростки >20 кг:
Стартовая доза: внутривенно: 55 мл/ м 2 (обеспечивает фенилацетат натрия 5,5 г/м2 и бензоат натрия 5,5 г/м2), вводимых в течение 90–120 минут.
Поддерживающая доза:
Непрерывная внутривенная инфузия: 55 мл/ м2 в виде поддерживающей инфузии в течение 24 часов (обеспечивает 5,5 г/м2 фенилацетата натрия и 5,5 г/м2 бензоата натрия в течение 24 часов);
Длительность: пожизненно.
|
А[29-32] |
| Карглуминовая кислота* | Доза от 100 до 250 мг/кг/сутки перорально 3 раза в день, длительность курса до нормализации уровня аммиака в крови | А[33,34] | |
| Фенилбутират натрия* |
Младенцы и дети <20 кг: Перорально: от 450 до 600 мг/кг/день во время еды/кормления от 3 до 6 раз в день; максимальная суточная доза: 20 г/сут. Длительность курса: пожизнено.
Дети ≥20 кг: Перорально: от 9,9 до 13 г / м 2 /день в во время еды от 3 до 6 раз в день; максимальная суточная доза: 20 г / сут. Длительность курса: пожизнено. |
А[18-20] | |
| Фенилбутират глицерол* |
Доза: 4,5 до 11,2 мл/м2 /сут.
Пациентам в возрасте 2 лет и старше: назначать в 3 равных дозировках, округляя каждую до ближайших 0,5 мл
Пациентам младше 2 лет: Назначать в 3 или более равных дозировках, округляя каждую до ближайших 0,1 мл.
Общая суточная доза не должна превышать 17,5 мл
|
А[6-18] | |
|
Аминокислоты
|
Гидрохлорид аргинина* |
Стартовая доза вводится внутривенно в течение 90 минут в 10% растворе глюкозы из расчета 25–35 мл/кг:
Для пациентов с массой тела ≤20 кг составляет 200 мг/кг,. Для пациентов >20 кг стартовая доза составляет 4 г/м2. Поддерживающая доза вводится внутривенно: 1) При дефиците CPSI составляет для пациентов: с массой тела ≤20 кг – 200 мг/кг в сутки; с массой тела >20 кг – 4 г/м2 в сутки. 2) При дефиците ASS и ASL составляет для пациентов: с массой тела ≤20 кг 600 мг/кг в сутки; с массой тела >20 кг.12 г/м2 в сутки
Длительность курса: пожизненно.
|
A[35-37] |
| Цитруллин* |
При дефиците CPSI также назначают перорально дозы цитруллина для пациентов: с массой тела ≤20 кг – от 150 до 200 мг/кг в сутки; с массой тела >20 кг и от 3 до 4 г/м 2 в сутки. Длительность курса: пожизненно. |
А[38] | |
| Ирригитационные растворы | Растворы глюкозы |
Внутривенно.
Начальная доза 10 мг/кг/мин, дальнейшая доза корригируется под контролем уровня глюкозы в крови.
Длительность курса: до перевода пациента на энтеральное питание
|
В[39,40] |
| Парентеральное питание | Комплекс аминокислот |
Внутривенно, из расчета от 1,5 до 1,75 г/кг белка в день.
Длительность курса: до перевода пациента на энтеральное питание
|
В[39,40] |
* не зарегистрирован в РК
Перечень дополнительных лекарственных средств [39-40]:
| Фармакотерапевтическая группа | МНН лекарственного средства | Способ применения | Уровень доказательности |
| Парентеральное питание | Жировые эмульсии |
Внутривенно, при исключении дефекта окисления жирных кислот, расчет дозы:
недоношенных, доношенных новорожденных – 0,5-4 г/кг/сут, младенцы и дети – 0,5-3 г/кг/сут.
Длительность курса: до перевода пациента на энтеральное питание
|
В[39,40] |
4. Дальнейшее ведение [41]:
Мониторинг пациента должен быть направлен на выявление и предотвращение чрезмерного ограничения белка, дефицита незаменимых аминокислот и увеличения поступления аммиака в организм.
Мониторинг включает в себя отслеживание уровня аммиака и аминокислот в крови, показателей функции печени.
|
NB! Лекарства, которых следует избегать [41]: • Глюкокортикоиды усиливают катаболизм белков и не должны использоваться рутинно. • Вальпроевая кислота ингибирует синтез мочевины, что приводит к повышению уровня аммиака в сыворотке, поэтому данное лекарство не должно использоваться для лечения судорог. • Маннитол неэффективен при лечении отека головного мозга, вызванного гипераммониемией из-за НЦМ. • Гепатотоксичные препараты следует использовать с осторожностью. |
Госпитализация
Показания для плановой госпитализации: нет
Показания для экстренной госпитализации [21-23]:
Информация
Источники и литература
-
Протоколы заседаний Объединенной комиссии по качеству медицинских услуг МЗ РК, 2023
- 1) Ah Mew, N.; Simpson, K.L.; Gropman, A.L.; Lanpher, B.C.; Chapman, K.A.; Summar, M.L. Urea cycle disorders overview. In GeneReviews ®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 2017. 2) Brusilow SW, Maestri NE. Urea cycle disorders: diagnosis, pathophysiology, and therapy. Adv Pediatr 2016; 43:127. 3) Moammar H, Cheriyan G, Mathew R, Al-Sannaa N. Incidence and patterns of inborn errors of metabolism in the Eastern Province of Saudi Arabia, 1983-2008. Ann Saudi Med 2018; 30:271. 4) Batshaw ML, Tuchman M, Summar M, et al. A longitudinal study of urea cycle disorders. Mol Genet Metab 2019; 113:127. 5) Matsumoto, S.; Haberle, J.; Kido, J.; Mitsubuchi, H.; Endo, F.; Nakamura, K. Urea cycle disorders-update. J. Hum. Genet. 2019, 64, 833–847. 6) Burrage LC, Madan S, Li X, et al. Chronic liver disease and impaired hepatic glycogen metabolism in argininosuccinate lyase deficiency. JCI Insight 2020; 5. 7) Leonard JV, Morris AA. Urea cycle disorders. Semin Neonatol 2020; 7:27. 8) Maestri NE, Brusilow SW, Clissold DB, Bassett SS. Long-term treatment of girls with ornithine transcarbamylase deficiency. N Engl J Med 2016; 335:855. 9) Gardeitchik T, Humphrey M, Nation J, Boneh A. Early clinical manifestations and eating patterns in patients with urea cycle disorders. J Pediatr 2019; 161:328. 10) Summar ML, Dobbelaere D, Brusilow S, Lee B. Diagnosis, symptoms, frequency and mortality of 260 patients with urea cycle disorders from a 21-year, multicentre study of acute hyperammonaemic episodes. Acta Paediatr 2018; 97:1420. 11) Burton BK. Inborn errors of metabolism in infancy: a guide to diagnosis. Pediatrics 2018; 102:E69. 12) Summar M. Current strategies for the management of neonatal urea cycle disorders. J Pediatr 2019; 138:S30. 13) Tuchman M, Yudkoff M. Blood levels of ammonia and nitrogen scavenging amino acids in patients with inherited hyperammonemia. Mol Genet Metab 2018; 66:10. 14) Savy, N.; Brossier, D.; Brunel-Guitton, C.; Ducharme-Crevier, L.; Du Pont-Thibodeau, G.; Jouvet, P. Acute pediatric hyperammonemia: Current diagnosis and management strategies. Hepatic. Med. Evid. Res. 2018, 10, 105–115. 15) Singh RH. Nutritional management of patients with urea cycle disorders. J Inherit Metab Dis 2017; 30:880. 16) Raina, R.; Bedoyan, J.K.; Lichter-Konecki, U.; Jouvet, P.; Picca, S.; Mew, N.A.; Machado, M.C.; Chakraborty, R.; Vemuganti, M.; Grewal, M.K.; et al. Consensus guidelines for management of hyperammonaemia in paediatric patients receiving continuous kidney replacement therapy. Nat. Rev. Nephrol 2020, 16, 471–482. 17) FDA news release. FDA approves new drug for the chronic management of some urea cycle disorders. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm337639.htm (Accessed on February 19, 2013). 18) Ravicti (glycerol phenylbutyrate) prescribing information. https://www.ravicti.com//files/RAVICTI_Prescribing_Information.pdf (Accessed on February 19, 2013). 19) Berry SA, Longo N, Diaz GA, et al. Safety and efficacy of glycerol phenylbutyrate for management of urea cycle disorders in patients aged 2months to 2years. Mol Genet Metab 2017; 122:46. 20) Maestri NE, Clissold D, Brusilow SW. Neonatal onset ornithine transcarbamylase deficiency: A retrospective analysis. J Pediatr 2018; 134:268. 21) Msall M, Batshaw ML, Suss R, et al. Neurologic outcome in children with inborn errors of urea synthesis. Outcome of urea-cycle enzymopathies. N Engl J Med 2016; 310:1500. 22) Jiang Y, Almannai M, Sutton VR, et al. Quantitation of phenylbutyrate metabolites by UPLC-MS/MS demonstrates inverse correlation of phenylacetate:phenylacetylglutamine ratio with plasma glutamine levels. Mol Genet Metab 2017; 122:39. 23) Fiati Kenston, S.S.; Song, X.; Li, Z.; Zhao, J. Mechanistic insight, diagnosis, and treatment of ammonia-induced hepatic encephalopathy. J. Gastroenterol Hepatol. 2019, 34, 31–39. 24) Walker V. Ammonia toxicity and its prevention in inherited defects of the urea cycle. Diabetes Obes Metab 2019; 11:823. 25) Singh RH. Nutritional management of patients with urea cycle disorders. J Inherit Metab Dis 2017; 30:880. 26) Summar M. Current strategies for the management of neonatal urea cycle disorders. J Pediatr 2017; 138:S30. 27) Darmaun D, Welch S, Rini A, et al. Phenylbutyrate-induced glutamine depletion in humans: effect on leucine metabolism. Am J Physiol 2018; 274:E801. 28) Green TP, Marchessault RP, Freese DK. Disposition of sodium benzoate in newborn infants with hyperammonemia. J Pediatr 2019; 102:785. 29) Brusilow SW, Valle DL, Batshaw M. New pathways of nitrogen excretion in inborn errors of urea synthesis. Lancet 2019; 2:452. 30) Praphanphoj V, Boyadjiev SA, Waber LJ, et al. Three cases of intravenous sodium benzoate and sodium phenylacetate toxicity occurring in the treatment of acute hyperammonaemia. J Inherit Metab Dis 2015; 23:129. 31) Thompson CA. Carglumic acid approved to treat genetic hyperammonemia. Am J Health Syst Pharm 2018; 67:690. 32) Daniotti M, la Marca G, Fiorini P, Filippi L. New developments in the treatment of hyperammonemia: emerging use of carglumic acid. Int J Gen Med 2018; 4:21. 33) Brusilow SW, Batshaw ML. Arginine therapy of argininosuccinase deficiency. Lancet 2019; 1:124. 34) Batshaw ML, Brusilow S, Waber L, et al. Treatment of inborn errors of urea synthesis: activation of alternative pathways of waste nitrogen synthesis and excretion. N Engl J Med 2019; 306:1387. 35) Lee B, Yu H, Jahoor F, et al. In vivo urea cycle flux distinguishes and correlates with phenotypic severity in disorders of the urea cycle. Proc Natl Acad Sci U S A 2018; 97:8021. 36) Tanaka K, Nakamura K, Matsumoto S, et al. Citrulline for urea cycle disorders in Japan. Pediatr Int 2017; 59:422. 37) Waisbren, S.E.; Gropman, A.L.; Batshaw, M.L. Improving long term outcomes in urea cycle disorders-report from the Urea Cycle Disorders Consortium. J. Inherit. Metab. Dis. 2016, 39, 573–584. 38) Morgan TM, Schlegel C, Edwards KM, et al. Vaccines are not associated with metabolic events in children with urea cycle disorders. Pediatrics 2017; 127:e1147. 39) Klein NP, Aukes L, Lee J, et al. Evaluation of immunization rates and safety among children with inborn errors of metabolism. Pediatrics 2019; 127:e1139. 40) Liotta EM, Romanova AL, Lizza BD, et al. Osmotic shifts, cerebraledema, and neurologic deterioration in severe hepatic encephalopathy. Crit Care Med. 2018;46(2):280–289.Singh RH. Nutritional management of patients with urea cycle disorders. J Inherit Metab Dis 2016; 30:880. 41) Singh RH. Nutritional management of patients with urea cycle disorders. J Inherit Metab Dis 2016; 30:880.
Информация
Список разработчиков протокола с указание квалификационных данных:
1) Абентаева Ботакоз Абубакировна – кандидат медицинских наук, доктор PhD, заведующая отделением реанимации и интенсивной терапии Корпоративного фонда «University Medical Center»;
2) Чарипова Бибигуль Толегеновна – доктор PhD, неонатолог, отделения реанимации и интенсивной терапии Корпоративного фонда «University Medical Center»;
3) Баянова Миргуль Файзуллиевна – кандидат медицинских наук, генетик, заведующая отделением клинико-генетической диагностики Корпоративного фонда «University Medical Center»;
4) Камзина Айгерим Бахытбековна – заведующая отделением интенсивной терапии новорожденных АО «Научный центр акушерства, гинекологии и перинатологии»;
6) Камиева Раушан Токтажановна – неонатолог, отделения реанимации и интенсивной терапии Корпоративного фонда «University Medical Center»;
7) Тлеугалиева Жанна Куандыккызы – неонатолог, отделения реанимации и интенсивной терапии Корпоративного фонда «University Medical Center»;
8) Жетимкаринова Гаухар Ерлановна – клинический фармаколог Корпоративного фонда «University Medical Center»;
Указание на отсутствие конфликта интересов: нет.
Рецензент: Жубанышева Карлыгаш Биржановна – кандидат медицинских наук, заведующая кафедрой неонатологии НУО «Казахстанско-Российский Медицинский университет», неонатолог-реаниматолог, высшая категория, профессор, Президент ассоциации неонатологов и специалистов детской медицины.
Указание условий пересмотра протокола: пересмотр не реже 1 раза в 5 лет и не чаще 1 раза в 3 года при наличии новых методов диагностики и лечения с уровнем доказательности.
Прикреплённые файлы
Внимание!
- Занимаясь самолечением, вы можете нанести непоправимый вред своему здоровью.
- Информация, размещенная на сайте MedElement и в мобильных приложениях "MedElement (МедЭлемент)", "Lekar Pro", "Dariger Pro", "Заболевания: справочник терапевта", не может и не должна заменять очную консультацию врача. Обязательно обращайтесь в медицинские учреждения при наличии каких-либо заболеваний или беспокоящих вас симптомов.
- Выбор лекарственных средств и их дозировки, должен быть оговорен со специалистом. Только врач может назначить нужное лекарство и его дозировку с учетом заболевания и состояния организма больного.
- Сайт MedElement и мобильные приложения "MedElement (МедЭлемент)", "Lekar Pro", "Dariger Pro", "Заболевания: справочник терапевта" являются исключительно информационно-справочными ресурсами. Информация, размещенная на данном сайте, не должна использоваться для самовольного изменения предписаний врача.
- Редакция MedElement не несет ответственности за какой-либо ущерб здоровью или материальный ущерб, возникший в результате использования данного сайта.