Болезнь Гентингтона
![]() Версия: Клинические протоколы МЗ РК - 2024 (Казахстан)
Версия: Клинические протоколы МЗ РК - 2024 (Казахстан)
Общая информация
Краткое описание
Одобрено
КЛИНИЧЕСКИЙ ПРОТОКОЛ ДИАГНОСТИКИ И ЛЕЧЕНИЯ
БОЛЕЗНЬ ГЕНТИНГТОНА
Болезнь Гентингтона (БГ) — это редкое прогрессирующее нейродегенеративное заболевание центральной нервной системы с аутосомно-доминантным типом наследования мутации, приводящей к увеличению числа тринуклеотидных повторов CAG цитозин-аденин-гуанин в гене HTT в 4-ой хромосоме [1-6].
ВВОДНАЯ ЧАСТЬ
Код(ы) МКБ:
| МКБ-10 | |
|
G10
|
Болезнь Гентингтона |
| МКБ-11 | |
|
8A01.10
|
Болезнь Гентингтона |
| 6D85.1 | Деменция вследствие болезни Гентингтона |
Дата разработки и пересмотра протокола: 2024 год.
Сокращения, используемые в протоколе:
|
АЛТ
|
Аланинаминотрансфераза |
| АСЛО | Антистрептолизин О |
| АСТ | Аспартатаминотрансфераза |
| АТА | Атаксия телеангиэктазия |
|
БГ
|
Болезнь Гентингтона |
| ВИЧ | Вирус иммунодефицита человека |
| ДНК | Дезоксирибонуклеиновая кислота |
| ИМТ | Индекс массы тела |
|
КФК
|
Креатинфосфокиназа |
| МРТ | Магнитно-резонансная томография |
| МСЭК | Медико-социальная экспертная комиссия |
|
ОАК
|
Общий анализ крови |
| ОКР | Обсессивно-компульсивное расстройство |
| РКИ | Рандомизированные клинические исследования |
| СИОЗС | Селективные ингибиторы обратного захвата серотонина |
|
СКВ
|
Системная красная волчанка |
| СРБ | С-реактивный белок |
| ЭЭГ | Электроэнцефалография |
| CAG | Цитозин-аденин-гуанин |
| COVID-19 | Коронавирусная болезнь 2019 |
|
HD
|
Huntington disease (Болезнь Гентингтона) |
| HDL | Huntington disease-like syndromes – синдромы, подобные болезни Гентингтона |
| HTT | Ген Huntingtin – ген, отвечающий за выработку белка хантингтина |
| SCA | Spinocerebellar Ataxia – Спиноцеребеллярная атаксия |
| TFC | Total Functional Capacity - Оценка общей функциональной способности |
Пользователи протокола: неврологи, терапевты, педиатры, врачи общей практики.
Категория пациентов: взрослые, дети.
Шкала уровня доказательности:
|
Класс (уровень) доказательности)
|
Виды научных исследований |
| А | Высококачественный мета-анализ, систематический обзор РКИ или крупное РКИ с очень низкой вероятностью (++) систематической ошибки, результаты которых могут быть распространены на соответствующую популяцию. |
| В | Высококачественный (++) систематический обзор когортных или исследований случай-контроль или Высококачественное (++) когортное или исследование случай-контроль с очень низким риском систематической ошибки или РКИ с не высоким (+) риском систематической ошибки, результаты которых могут быть распространены на соответствующую популяцию. |
| С | Когортное или исследование случай-контроль или контролируемое исследование без рандомизации с не высоким риском систематической ошибки (+), результаты которых могут быть распространены на соответствующую популяцию или РКИ с очень низким или невысоким риском систематической ошибки (++ или +), результаты которых не могут быть непосредственно распространены на соответствующую популяцию. |
| D | Описание серии случаев или неконтролируемое исследование или мнение экспертов. |
Классификация
Классификация
По стадии прогрессирования
БГ делят на [7]:
Доклиническая стадия (имеется генетическое подтверждение, но еще нет симптомов заболевания) – отсутствуют двигательные, когнитивные симптомы; могут присутствовать изменения на МРТ; симптоматическое лечение не требуется.
Продромальная стадия (генетически подтвержденная БГ или без генетического подтверждения) – незначительные моторные и когнитивные клинические симптомы, значительно не влияющие на качество жизни; возможна депрессия, изменения на МРТ; симптоматическая терапия может потребоваться в некоторых случаях (например, антидепрессанты).
Клинически манифестирующая стадия (генетически подтвержденная БГ или без генетического подтверждения) – наличие моторных и когнитивных нарушений, влияющих на качество жизни пациента; требуется медикаментозная терапия.
По возрасту дебюта БГ разделяют на [8,9]:
ювенильная БГ (Вестфальский вариант) – дебют заболевания до 20 лет;
с ранним дебютом – дебют в 20-29 лет;
обычный дебют – дебют в возрасте 30-59 лет (средний возраст дебюта 35-45 лет);
поздний дебют – дебют в возрасте 60 лет и старше.
По тяжести функционального нарушения БГ делится на стадии [8,10] (Total Functional Capacity - Оценка общей функциональной способности - Приложение 1):
стадия I - TFC 11-13 баллов;
стадия II - TFC 7-10 баллов;
стадия III - TFC 3-6 баллов;
стадия IV - TFC 1-2 баллов;
стадия V - TFC 0 баллов.
По преимуществу тех или иных моторных проявлений [11]:
преимущественно хореические (21,55% случаев);
гипокинетически-ригидные (22,15% случаев);
смешанно-двигательные (56,30%) случаев.
Клиническая картина
Cимптомы, течение
Клиническая картина заболевания
Проявления включают двигательные, когнитивные и личностные изменения.
При Вестфальском варианте (ювенильная БГ) выражены дистония и паркинсонизм [1]. При БГ проявляются либо все эти группы симптомов, либо 1-2, либо 1 – в самом начале заболевания. У детей достаточно часто выявляется эпилепсия.
Диагностика
МЕТОДЫ, ПОДХОДЫ И ПРОЦЕДУРЫ ДИАГНОСТИКИ
Диагностические критерии.
Жалобы
(чаще со слов не столько пациента, сколько со стороны его близких, поскольку из-за сниженной критики сами пациенты могут не обращать внимания на свои симптомы [12,13]):
- непроизвольные движения, беспокойство, «подергивания»,
- нарушение походки, баланса, падения,
- неуклюжесть, неряшливость (роняют, разбивают предметы, еду, разливают напитки),
- скованность, замедленность движений, - нарушение речи,
- скрежетание зубами по ночам,
- изменение почерка,
- затруднение глотания, поперхивания,
- снижение памяти, внимания,
- эмоциональная лабильность, раздражительность,
- агрессия,
- расторможенность, эйфория,
- сниженный фон настроения,
- бред и навязчивые действия,
- сексуальная распущенность,
- галлюцинации,
- суицидальные высказывания, действия,
- снижение веса,
- повышенное потоотделение,
- недержание мочи, неудержание мочи, императивные позывы, никтурия.
При ювенильной БГ клиническая картина, а следовательно, и жалобы отличаются от взрослой формы БГ [14]. Жалобы звучат со стороны родителей и близких ребенка:
- потеря самоконтроля поведения,
- импульсивность,
- эмоциональная лабильность,
- раздражительность,
- вспышки гнева,
- тревога/чувство вины,
- повторение одной и той же мысли или идей (настойчивость),
- трудности с изучением нового материала,
- невнятная речь,
- бруксизм,
- затруднение письма,
- слюнотечение,
- затруднения при глотании,
- неуклюжесть,
- скованность в конечностях,
- ходьба на носочках,
- перекрест нижних конечностей при ходьбе,
- нарушение координации,
- потеря ранее приобретенных навыков (бросание мяча, езда на велосипеде и др.),
- непроизвольные движения,
- приступы потери сознания (эпилепсия является распространенным симптомом БГ у детей, встречается у 90% пациентов с началом в первые 5 лет жизни и у 80% детей с началом в возрасте от 6 до 10 лет [15,16],
- недержание мочи, неудержание мочи, императивные позывы, никтурия.
Анамнез:
Семейный анамнез – наличие похожих симптомов у одного из родителей или других близких родственников или наличие генетически подтвержденной БГ у ближайших родственников (не обязательно, так как не всегда удается проследить у всех родственников, кроме того, есть вероятность спонтанных мутаций, что наблюдается у 1-4% пациентов с БГ) [17].
Постепенное прогрессирование симптомов на протяжении нескольких месяцев – лет.
Известно, что при БГ наблюдается антиципация – феномен, при котором в последующих поколениях наблюдается увеличение тяжести заболевания или уменьшение возраста начала заболевания. Ожидание гораздо чаще возникает при передаче мутировавшего аллеля от отца [18]. Большие расширения (т. е. увеличение размера аллелей >7 CAG-повторов) происходят почти исключительно за счет отцовской передачи. Чаще всего дети с ювенильным началом заболевания наследуют расширенный аллель от отцов, хотя иногда они наследуют его и от матерей.
Физикальное обследование:
Необходимо отметить, что БГ является заболеванием с широким клиническим разнообразием. При осмотре у пациентов могут определяться следующие нарушения:
- хорея (является ключевым признаком БГ и определяющим симптомом на момент постановки диагноза; в дебюте заболевания движения легкие и могут быть ошибочно приняты за беспокойство; пациенты могут не осознавать своих движений и включать хорею в целенаправленные действия (скрещивание и разгибание ног, поправка очков, потирание подбородка и т.д.) - феномен, получивший название «паракинезия»; более характерна для взрослых пациентов);
- ригидность (более характерна для ювенильной формы БГ; иногда у взрослых пациентов в более продвинутых стадиях заболевания хореический гиперкинез может смениться ригидностью и брадикинезией);
- брадикинезия (более характерна для ювенильной формы БГ; иногда у взрослых пациентов в более продвинутых стадиях заболевания хореический гиперкинез может смениться ригидностью и брадикинезией);
- дистония (чаще у детей);
- атаксия (чаще у детей и пациентов молодого возраста);
- миоклонии (чаще у детей; могут быть проявлением миоклонических эпилептических приступов);
- сниженный мышечный тонус (более характерна для взрослых пациентов; сниженный тонус в сочетании с гиперрефлексией может быть ранним признаком заболевания);
- гиперрефлексия (не всегда);
- нарушение плавного слежения, замедленные саккады (пациенты испытывают затруднения с инициацией саккад, из-за чего используют вспомогательные техники для их выполнения: использование толчков головой или моргание, рефиксация);
- снижение когнитивных функций (когнитивное тестирование у взрослых и детей выявляет разной степени когнитивные нарушения; у детей симптомы похожи на синдром дефицита внимания и гиперактивности);
- акатизия (синдром, характеризующийся неприятными ощущениями «внутреннего» беспокойства, проявляющегося в неспособности сидеть на месте.);
- дисграфия;
- дисфагия;
- дизартрия;
- депрессия;
- тревога;
- разражительность;
- обсессивно-компульсивное поведение;
- агрессивное поведение;
- паранойя, бред.
Лабораторные исследования
Основные:
- Генетическое тестирование
БГ вызывается экспансией тринуклеотида цитозин-аденин-гуанина (CAG) в гене HTT (также известном как ген HD), который кодирует белок хантингтин, что приводит к расширению полиглутаминового тракта.
Метод тестирования – фрагментарный анализ. Необходимо тестировать симптомных пациентов, а также асимптомных родственников старше 18 лет. Профилактическое тестирование лиц, не достигших 18 лет не рекомендуется. Возможно применение пренатального тестирования и преимплантационного генетического тестирования. На начальных стадиях заболевания при подозрении на ювенильную форму рекомендуется провести тщательный неврологический осмотр, нейропсихологическое обследование, МРТ головного мозга с повтором комплекса обследования через 6-12 месяцев. При ухудшении по какому-либо из этих параметров целесообразно проведение генетического тестирования.
Основным фактором, определяющим возраст начала заболевания, является количество CAG-повторов в гене HTT. Нормальное количество повторов — 26 или меньше. При повторах 27-35 симптомы не развиваются, но у следующего поколения существует небольшой риск развития экспансии, которая может входить или не входить в диапазон, вызывающий заболевание. Повторы 36-39 не полностью пенетрантные; у отдельных людей могут развиваться симптомы, но обычно они появляются в позднем возрасте. Когда количество повторов 40 и более, заболевание является полностью пенетрантным и возникают симптомы заболевания. При раннем появлении симптомов (у детей и в возрасте моложе 30 лет), как правило, наблюдается наибольшее увеличение количества повторов, тогда как начало в позднем возрасте коррелирует с меньшим увеличением количества повторов. Скорость прогрессирования заболевания также обратно пропорциональна размеру повторов [19]. При очень ранней форме ювенильной болезни Гентингтона число CAG-повторов превышает 80 [20,21].
После сообщения положительного результата ДНК-тестирования необходимо предоставить пациенту более подробную информацию относительно клинических проявлений БГ, социальных и психологических последствий выставляемого диагноза, подходов к планированию семьи, доступных на текущий момент методов лечения заболевания.
Основные:
- МРТ головного мозга
Аксиальная МРТ боковых желудочков демонстрирует атрофию хвостатого ядра, определяемую потерей нормального выпячивания головки хвостатого ядра в боковой желудочек на поздней стадии БГ. Атрофия хвостатого ядра, количественно определяемая с помощью простого анализа линейных измерений хвостатого ядра, коррелирует с изменением когнитивной функции [19]. Также на МРТ определяется атрофия скорлупы. На МРТ может выявляться прогрессирующая атрофия серого и белого вещества за много лет до предполагаемого начала заболевания [22]. МРТ играет второстепенную роль после генетического анализа.
Дополнительные:
- Видео-ЭЭГ-мониторинг
Консультация профильных специалистов:
На стадии установки диагноза для проведения дифференциальной диагностики:
- ревматолог;
- инфекционист;
- эндокринолог;
- психиатр.
При генетически установленном диагнозе БГ:
- врач-генетик;
- реабилитолог;
- психиатр;
- логопед;
- хирург;
- стоматолог [24];
- диетолог;
- гастроэнтеролог;
- уролог [25];
- психолог;
- социальные работники.
Диагностический алгоритм [26].
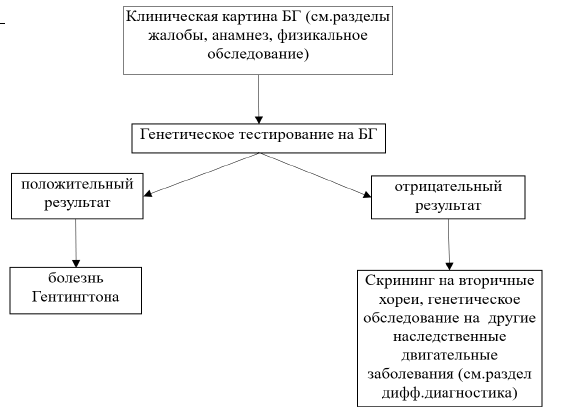
Рисунок 1. Алгоритм диагностики БГ.
Дифференциальный диагноз
Дифференциальный диагноз и обоснование дополнительных исследований.
Таблица 1 – Дифференциальный диагноз БГ.
|
Диагноз
|
Обоснование для дифференциальной диагностики | Обследования | Критерии исключения диагноза |
| Другие аутосомно-доминантные двигательные расстройства, схожие с БГ | |||
| Экспансия повторов C9ORF72 | Фенотипически может проявляться как БГ. У пациентов могут наблюдаться хорея, дистония, атаксия, паркинсонизм, миоклонус и пирамидные признаки. | Генетическое тестирование | Мутации в гене C9ORF |
| Синдромы, подобные БГ (Huntington disease-like syndromes - HDL syndromes) | HDL обычно проявляются как БГ, также возможны эпилептические приступы. Это вызвано вставками октапептидных повторов в ген PRNP на хромосоме 20, кодирующий прионный белок. | Генетическое тестирование | Вставки октапептидных повторов в ген PRNP на хромосоме 20 |
| Доброкачественная наследственная хорея | Проявляется хореей в младенчестве, которая может улучшиться с возрастом. Несмотря на свое название, это заболевание не всегда доброкачественное. У многих пациентов также наблюдаются трудности в обучении или поведении, задержка двигательных показателей, дизартрия, аксиальная дистония, нарушения походки, тики, тремор или миоклонические подергивания. | Генетическое тестирование |
Аутосомно-доминантные мутации гена NKX2-1 (также известного как ген TITF1) на хромосоме 14q.
Фенотипы, сходные с доброкачественной наследственной хореей, связанной с геном NKX2-1, были описаны с мутациями в других генах, включая ADCY5, PDE10A, GNAO1 и SLC16A2. Поскольку ген NKX2-1 также отвечает за развитие легких и щитовидной железы, вовлечение этих органов часто сосуществует и может быть диагностическим ключом, отсюда и термин «синдром мозг-легкие-щитовидная железа».
|
| Дентаторубральная паллидолуизовая атрофия (DRPLA) | У взрослых DRPLA вызывает атаксию, хореоатетоз и снижение когнитивных функций и может имитировать БГ. В юношеских случаях встречается миоклоническая эпилепсия, которая часто встречается в юношеских случаях. | Генетическое тестирование | Дентаторубральная паллидолуизовая атрофия — DRPLA — редкое заболевание, вызванное экспансией триплета CAG в гене ATN1, кодирующем атрофин-1. Расширение может привести к ожиданию передачи по отцовской линии. ДРПЛА особенно распространена в Японии, но встречается и в других этнических группах. Возраст начала варьируется в широких пределах. |
| Спиноцеребеллярная атаксия (SCA) 1,2,3,17 типов | Группа гетерогенных расстройств, характеризующихся прогрессирующей мозжечковой атаксией, часто сочетающейся с другими неврологическими признаками, такими как офтальмоплегия, пирамидные или экстрапирамидные симптомы, глубокая потеря чувствительности и деменция. Начало может быть в детстве или пожилом возрасте, но обычно происходит в возрасте от 20 до 40 лет. Типы SCA 1, 2, 3 и 17 чаще, чем другие типы SCA, вызывают хорею. | Генетическое тестирование | Генетически подтвержденные варианты SCA |
| Мутации ADCY5 и PDE10A | Данные мутации вызывают широкий спектр гиперкинетических двигательных расстройств, возникающих в детстве, иногда называемых семейной дискинезией с миокимией лица. Хорея часто является основным признаком, также у пациентов могут наблюдаться миокимии лица, дистония и миоклонус. У некоторых пациентов могут возникать эпизодические приступы, прежде чем двигательное расстройство станет постоянным. |
Генетическое тестирование.
МРТ головного мозга.
|
Мутация генов ADCY5 или PDE10A.
Появление симптомов в первые годы жизни; выраженные подергивания лица; пароксизмальный характер гиперкинезов с обострением ночью или при пробуждении.
На МРТ головного мозга гиперинтенсивные двусторонние поражения полосатого тела.
|
| Нейроферритинопатия (NBIA) | Редкий прогрессирующий синдром, который вызывает паркинсонизм, дистонию, снижение когнитивных функций и другие неврологические нарушения, характеризуется появлением во взрослом возрасте экстрапирамидных проявлений, таких как хорея, дистония или паркинсонизм, которые обычно носят асимметричный характер, а также когнитивныхх нарушений. Также могут наблюдаться дизартрия, спастичность и мозжечковые симптомы. |
Генетическое тестирование.
МРТ головного мозга.
Анализ крови на ферритин.
|
Мутации гена FTL, кодирующего легкую цепь ферритина.
На МРТ аномальное отложение железа, кистозные изменения и некроз в базальных ганглиях.
|
| Аутосомно-рецессивные двигательные расстройства, схожие с БГ | |||
| Хорея-акантоцитоз | К клиническим проявлениям хореи-акантоцитоза относятся хорея, дистония, тики, паркинсонизм, нарушения движений глаз, поведенческие изменения и снижение когнитивных функций, судороги. Фенотипически может быть сходен с БГ. Средний возраст начала заболевания составляет 30 лет, заболевание неуклонно прогрессирует, смерть наступает в течение 15 лет после начала. |
Генетическое тестирование.
Анализ крови (мазок) + КФК
|
Мутация гена VPS13A на хромосоме 9, который кодирует белок хореин.
В мазках периферической крови обычно присутствуют акантоциты, но их отсутствие не исключает диагноза.
Периферическая нейропатия с дистальной амиотрофией.
Повышение уровня креатинкиназы в сыворотке.
|
| Болезнь Вильсона |
Хорея является редким симптомом болезни Вильсона, но это состояние нельзя упускать из виду, поскольку это излечимое заболевание. Болезнь Вильсона следует предполагать у всех пациентов с двигательными расстройствами в возрасте до 40 лет.
Неврологические проявления болезни Вильсона включают тремор, дизартрию, атаксию, дистонию, паркинсонизм и слюнотечение, также могут включать тики, миоклонус, хорею, дисфонию, судороги, а также поведенческие и когнитивные изменения.
|
Генетическое тестирование.
Анализ на медь и церуллоплазмин.
Офтальмологический осмотр на щелевой лампе.
Анализ крови на церуллоплазмин, медь.
Суточный анализ мочи на медь.
МРТ головного мозга.
|
Мутация гена ATP7B, приводящая к неуклонному мультисистемному накоплению меди.
Выявление колец Кайзера-Флейшера.
Низкий уровень церуллоплазмина (может быть в норме в 5–15% процентах случаев), низкий уровень меди в сыворотке, повышенная суточную экскрецию меди с мочой и отклонения от нормы функциональных проб печени.
МРТ головного мозга - гиперинтенсивность в базальных ганглиях на Т2-взвешенных изображениях или аномалии сигналов в среднем мозге («лицо гигантской панды»).
|
| Нейродегенерация, связанная с пантотенаткиназой (PKAN) - болезнь Халлервордена-Шпатца | Начало заболевания приходится на первое десятилетие, обычно дистония походки развивается в генерализованную дистонию, дизартрию, паркинсонизм, спастичность, пигментную дегенерацию сетчатки, деменцию и поведенческие изменения. |
Генетическое тестирование.
МРТ головного мозга.
|
Мутации в гене, кодирующем пантотенат-киназу 2 (PANK2), приводящие к накоплению железа в базальных ганглиях, больше выраженному во внутреннем бледном шаре.
МРТ головного мозга - «тигровый глаз», с центральным фокусом повышенной интенсивности сигнала Т2 в медиальном бледном шаре, окруженном зоной пониженного сигнала.
|
| Атаксия-телеангиэктазия | Заболевание обычно начинается в возрасте от одного до четырех лет с прогрессирующей мозжечковой атаксией, со временем присоединяются глазодвигательная апраксия, хорея, дистония, миоклонус и периферическая нейропатия. Сопутствующие признаки: телеангиэктазии конъюнктивы или лица, иммунодефицит с рецидивирующими синуситами или легочными инфекциями, повышенную заболеваемость злокачественными новообразованиями, чувствительность к радиации и сахарный диабет, вызванный резистентностью к инсулину. | Генетическое тестирование. |
Мутации в гене мутированной атаксии телеангиэктазии (АТМ) на хромосоме 11; продукт гена ATM участвует в обнаружении повреждений ДНК.
Телеангиэктазии конъюнктивы или лица.
Снижение сывороточного иммуноглобулина А, повышение сывороточного альфа-фетопротеина.
|
| Вторичные хореи | |||
| Структурные изменения в области базальных ганглиев (опухоль, инсульт, энцефалит) | Очаговое поражение головного мозга в области базальных ганглиев может вызвать появление симптомов хореи, баллизма | МРТ головного мозга, в том числе с контрастированием |
Острое или подострое возникновение симптомов.
Очаговые структурные изменения в области базальных ганглиев на МРТ.
|
| Хорея Сиденгама | Наиболее частая причина хореи в детском возрасте. Это одно из клинических проявлений острой ревматической лихорадки, негнойного осложнения стрептококковой инфекции группы А. Обычно возникает у детей в возрасте от 5 до 15 лет, хотя он также был описан и у взрослых. Хорея обычно возникает через 4-8 недель после инфекции, реже позже – од 8 месяцев. Также могут наблюдаться тики, дизартрия, гипотония, мышечная слабость, нарушение походки и вокализация. Также заболевание ассоциируется с психологическими и психиатрическими проявлениями, которые могут предшествовать возникновению хореи, включая эмоциональную лабильность, раздражительность, невнимательность, гиперактивность, обсессивно-компульсивное поведение, тревога, депрессия. Приступы хореи обычно длятся от двух до девяти месяцев, после чего полностью спонтанно разрешаются. Однако у значительной части пациентов хорея сохраняется через два года или наблюдается рецидив. | Титр антистрептолизина О. | Титр АСЛО имеет ограниченное применение у пациентов с хореей Сиденгама, поскольку титры обычно достигают пика до появления симптомов, а дети без ревматизма или хореи часто имеют низкие положительные титры АСЛО. |
| Паранеопластическая хорея. |
Хорея является редким паранеопластическим синдромом, обычно связанным с антителами к CV2/коллапсин-чувствительному медиаторному белку 5 (CRMP5) в контексте мелкоклеточной карциномы легкого. У большинства этих пациентов также наблюдаются другие неврологические признаки и симптомы, такие как потеря зрения, невропатия, атаксия или лимбический энцефалит. Имеются данные также о хорее при других видах рака (рак молочной железы, тимома, лимфома, почечно-клеточный рак и рак яичек) и при наличии других типов антител (анти-Hu, анти-Ri, анти-Yo).
Паранеопластический энцефалит, связанный с антителами к рецептору N-метил-D-аспартата (NMDA), часто проявляется аномальными движениями, включая хорею, стереотипии, дистонию и миоклонус, и обычно сочетается с другими неврологическими симптомами, такими как поведенческие или психиатрические проявления, атаксия и судороги. NMDA-энцефалит может быть связан с тератомой яичника или может быть аутоиммунным, без основной опухоли.
Энцефалит, вызванный антителами к рецепторам инактивированной глиомы 1, богатой лейцином (LGI1), контактин-ассоциированным белком 2 (Caspr2), Hu или рецепторам гамма-аминомасляной кислоты типа B (GABA-B), также может проявляться двусторонней или односторонней хореей с либо паранеопластического, либо аутоиммунного происхождения.
|
Скрининг на паранеопластические антитела.
МРТ головного мозга.
|
Определение паранеопластических антител, первичного онкологического очага.
У большинства пациентов с паранеопластической хореей на МРТ наблюдаются аномальные сигналы в базальных ганглиях.
|
| Другие иммуноопосредованные хореи | Поражение центральной нервной системы, в том числе с проявлениями в виде хореи, может наблюдаться при СКВ, антифосфолипидном синдроме, при синдроме Бехчета, синдроме Шегрена, васкулите иммуноглобулина А (IgAV; пурпура Шенлейна-Геноха), узелковом полиартериите, первичном ангиите центральной нервной системы, целиакии и саркоидозе. |
Скрининг на системные заболевания.
Консультация ревматолога.
|
Лабораторно и клинически подтвержденные системные заболевания |
| Метаболические нарушения | Гипергликемия, гипогликемия, гипертиреоз, гипернатриемия, гипонатриемия, гипомагниемия, гипокальциемия, дефицит витамина В12, хроническая приобретенная гепатоцеребральная дегенерация могут вызывать развитие симптомов хореи, а также других двигательных расстройств, таких как тремор, паркинсонизм, миоклонус или дистония. |
Биохимический анализ крови.
МРТ головного мозга.
|
Подтвержденные метаболические нарушения по данным биохимических анализов.
На МРТ головного мозга стриатопатия в виде гиперинтенсивного сигнала на Т1-взвешенных изображениях в контралатеральном полосатом теле при нормальных диффузионно-взвешенных изображениях.
|
| Инфекционные заболевания | Многие инфекционные заболевания центральной нервной системы могут вызывать острую или подострую хорею. ВИЧ является наиболее частой причиной инфекционной хореи. Также симптомами хореи могут проявляться токсоплазмоз, сифилис, туберкулез, болезнь Лайма, паразитарные или грибковые инфекции, а также прионные заболевания могут вызывать хорею у некоторых пациентов. Хорея редко регистрировалась при вирусном энцефалите, в том числе в случаях COVID-19 и инфекции, вызванной вирусом простого герпеса. |
Анализ крови и ликвора на инфекции.
МРТ головного мозга.
|
Подтвержденные по результатам анализов и МРТ внутримозговые инфекции. |
| Воздействие токсинов | Интоксикация угарным газом, марганцем, таллием, толуолом, метанолом, цианидом или ртутью может привести к временным или постоянным двигательным расстройствам, включая хорею. Употребление алкоголя, амфетаминов, кокаина или героина, а также вдыхание клея также могут вызвать хорею. |
Анамнез.
Анализы.
МРТ головного мозга.
|
Данные анамнеза и обследования, подтверждающие токсическую природу гиперкинеза. |
| Хорея, вызванная лекарственными средствами | Некоторые лекарственные препараты при длительном применении могут вызывать двигательные нарушения (например, нейролептики и противорвотные средства, такие как метоклопрамид и прохлорперазин. Использование пероральных контрацептивов может привести к односторонней или генерализованной хорее, особенно у лиц с предшествующей СКВ или антифосфолипидным синдромом. Время между началом лечения и появлением аномальных движений может варьироваться от нескольких дней до нескольких лет. Также описана хорея при заместительной гормональной терапии у женщин в постменопаузе. | Анамнез. | Данные анамнеза и обследования, подтверждающие лекарственную природу гиперкинеза. |
Лечение (амбулатория)
ТАКТИКА ЛЕЧЕНИЯ НА АМБУЛАТОРНОМ УРОВНЕ
1. Немедикаментозная терапия
Диета
Потеря веса часто присутствует при ГБ, иногда до появления других симптомов [27]. Из-за повышенного метаболизма, гиперкинеза и проблем с жеванием и глотанием у пациентов может наблюдаться потеря веса. В связи с чем необходимо следить за полноценным питанием пациентов. Для определения возможных других причин потери веса следует учитывать такие факторы, как способность глотания, когнитивные изменения, поведение, настроение и общие функциональные способности (уровень C) [28].
По возможности следует поддерживать высокий индекс массы тела (ИМТ) в пределах нормальных значений и рекомендовать медицинское и/или социальное вмешательство, если непреднамеренная потеря веса превышает 10% за последние 3–6 месяцев или при ИМТ <20 и непреднамеренной потере веса на 5% в течение последних 3–6 месяцев. Назначаются высококалорийные и высокобелковые пищевые добавки под контролем диетолога/диетолога (уровень С) [29]. Средиземноморская диета может улучшить качество жизни и состав питательных веществ (уровень C) [30].
В случае начала лечения антидепрессантами и/или нейролептиками у пациентов со значительной потерей веса следует отдавать предпочтение лечению, вызывающему увеличение веса, в то время как следует избегать лечения, вызывающего потерю веса (эти эффекты могут варьироваться от одного пациента к другому) (уровень C) [31].
Крайне важно заблаговременное планирование ухода, предусмотрение альтернативных методов кормления, обсуждение их с родственниками и пациентами, которые все еще способны понимать преимущества и риски вмешательства. У пациентов с дисфагией необходима дополнительная нутритивная поддержка, при необходимости зондовое питание, наложение гастростомы [32].
Водно-электролитный баланс
Избыточная потливость присутствует на всех стадия БГ, она связаны с автономными нарушениями при заболевании. Во всех случаях потливости ухаживающим лицам необходимо следить за достаточным поступлением воды и электролитов. Кроме того, при чрезмерной потливости необходимо помнить о возможной инфекции или дисфункции щитовидной железы и обследовать пациента соответствующим образом.
Занятия с логопедом, улучшение глотания
У пациентов с БГ зачастую появляются проблемы с глотанием, речью, в связи с чем необходимой составляющей немедикаментозной терапии являются занятия с логопедом как можно раньше от начала появления нарушений, в дальнейшем необходимо проводить регулярную оценку нарушений глотания (уровень доказательности А) [33]. Необходимо обучение лиц, осуществляющих уход, важно, поскольку они часто контролируют режим еды, питья и глотания. В тяжелых случаях может понадобиться введение назогастрального зонда и наложения чрезкожной эндоскопической гастростомы [34].
Психологическая помощь
Хорея может усиливаться при тревоге и стрессе [35]. Обеспечение спокойной, предсказуемой и структурированной среды наряду с психологической консультативно-терапевтической помощью и когнитивно-поведенческой терапией может помочь решить эти проблемы. Родственникам и ухаживающим лицам необходимо проявлять суицидальную настороженность при малейшем подозрении обращаться за помощью специалистам. Суицидальные мысли или попытки часто встречаются при БГ независимо от стадии заболевания [36]. Особую бдительность необходимо проявлять в момент постановки диагноза и в дальнейшем, когда заболевание начинает влиять на повседневную жизнь. Профилактика самоубийств включает в себя лечение таких факторов риска, как депрессия, социальная изоляция и импульсивность [37].
Лечебная физкультура
Для улучшения баланса, ходьбы, навыков самообслуживания пациентам с БГ требуется физическая реабилитация в виде индивидуальных занятий с инструктором ЛФК [38]. Пациентам с БГ рекомендованы аэробные упражнения (уровень доказательности А), отдельно или в сочетании с тренировками с отягощениями для улучшения физической формы и двигательных функций, а также тренировку ходьбы под наблюдением (уровень доказательности А) [39].
Вспомогательные технологии
Пациентам с тяжелой хореей может быть полезно использование вспомогательного оборудования, такого как шлемы, мягкие кресла с откидной спинкой, низкие кровати, инвалидное кресло и другие приспособления [19].
Вследствие недержания мочи пациентам необходимы подгузники и урологические прокладки. При недержании кала пациентам могут оказаться необходимы калоприемники.
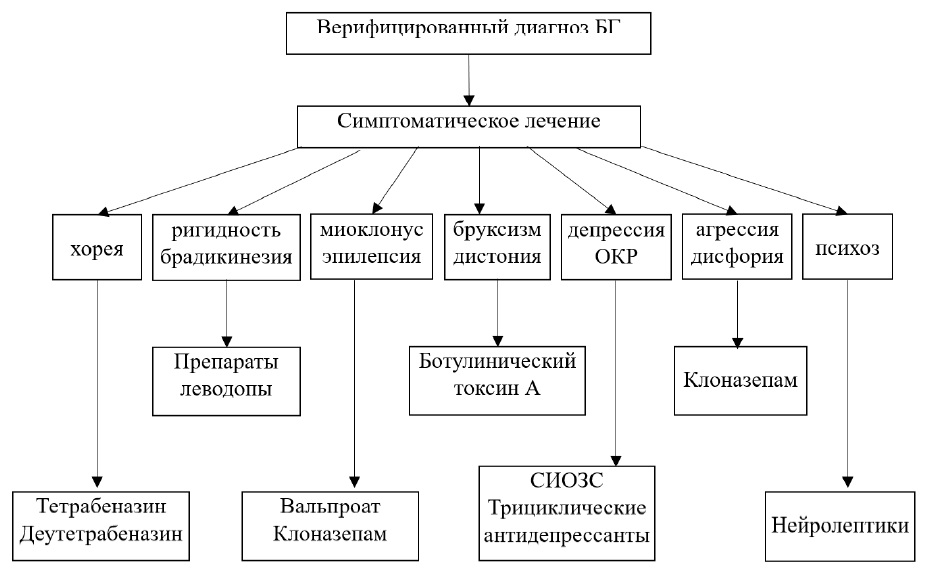
Рисунок 2. Алгоритм лечения БГ.
Симптоматическое лечение хореи.
Таблица 2 – Препараты для уменьшения проявления хореического гиперкинеза [19,32].
|
Фармакотерапевтическая группа
|
МНН | Способ применения | Уровень доказательности |
| Препараты 1 линии | |||
| Ингибитор везикулярного переносчика моноаминов (VMAT2), другие нейротропные средства | Тетрабеназин* |
Начальная дозировка 12,5 мг от 1 до 3 раз в сутки.
Далее доза может быть увеличена 1 раз в 3–4 дня на 12,5 мг до достижения оптимального терапевтического эффекта или до обнаружения побочных эффектов (седативный эффект, паркинсонизм, депрессия).
Максимальная суточная доза составляет 200 мг в сутки
|
А |
| Ингибитор везикулярного переносчика моноаминов (VMAT2), другие нейротропные средства | Деутетрабеназин* |
Начальная дозировка 6 мг внутрь.
Возможно увеличение суточной дозировки постепенно, каждые 7 дней на 6 мг.
Средняя рекомендуемая дозировка – 12 мг.
Максимальная суточная дозировка 36 мг.
|
А |
| Препараты 2 линии | |||
| Атипичный нейролептик | Оланзапин |
Стартовая дозировка: 5 мг внутрь.
Максимальная суточная дозировка: 30 мг.
|
В |
|
Атипичный нейролептик
|
Рисперидон | Стартовая дозировка: 0,5 мг 1 раз в день внутрь, при отсутствии положительного эффекта, возможно увеличить до 1 мг (кратность приема по 0,5 мг 2 раза в день). Максимальная суточная дозировка: 4–6 мг | В |
|
Бензодиазепины
|
Клоназепам | Стартовая дозировка: 0,5 мг внутрь ежедневно. Максимальная суточная дозировка 2-3 мг | С |
|
Типичный нейролептик
|
Галоперидол |
По 0,5-1 мг внутрь 1-2 раза в день ежедневно.
Максимальная суточная дозировка: 10 мг
|
С |
Таблица 2.1 – Препараты для уменьшения проявления хореического гиперкинеза в детском возрасте [54-58].
|
Фармакотерапевтическая группа
|
МНН | Способ применения | Уровень доказательности |
| Препараты 1 линии | |||
| Ингибитор везикулярного переносчика моноаминов (VMAT2), другие нейротропные средства | Тетрабеназин* | 12,5 мг от 1 до 3 раз в сутки. Далее доза может быть увеличена 1 раз в 3–4 дня на 12,5 мг до достижения оптимального терапевтического эффекта или до обнаружения побочных эффектов (седативный эффект, паркинсонизм, депрессия). Максимальная суточная доза составляет 200 мг в сутки | А |
| Ингибитор везикулярного переносчика моноаминов (VMAT2), другие нейротропные средства | Деутетрабеназин* |
Начальная дозировка 6 мг перорально у детей в возрасте от 6-16 лет. Возможно увеличение суточной дозировки постепенно, каждые 7 дней на 6 мг.
Средняя рекомендуемая дозировка – 12 мг.
Максимальная суточная дозировка 36 мг.
|
А |
| Препараты 2 линии | |||
| Атипичный нейролептик | Рисперидон | Детям принимать препарат дважды в день, перорально в суточной дозе: 2-12 лет - 0,5-2 мг; в возрасте 12-18 лет 1-4 мг. | В |
|
Бензодиазепины
|
Клоназепам |
Дети в возрасте 1-5 лет начальная доза не более 250 мкг/сут; дети в возрасте 5-12 лет - 500 мкг/сут, перорально. Поддерживающие суточные дозы для детей в возрасте до 1 года - 0.5-1 мг, 1-5 лет - 1-3 мг, 5-12 лет - 3-6 мг.
Подбор дозы индивидуален.
|
С |
| Типичный нейролептик | Галоперидол |
Начальная доза: 0,5 мг/день перорально в 2–3 приема. Поддерживающая доза: от 0,05 до 0,075 мг/кг/день |
С |
NB! В продвинутых стадиях у взрослых пациентов гиперкинез может смениться брадикинезией. В связи с чем может потребоваться коррекция терапии с отменой препаратов этой группы и необходимостью назначения препаратов следующей группы (табл. 3).
Таблица 3 – Препараты для лечения брадикинезии и акинетико-ригидного синдрома при БГ.
|
Фармакотерапевтическая группа
|
МНН | Дозировка | Уровень доказательности |
| Противопаркинсонический препарат. Дофаминергическое средство. | Леводопа 250/ Карбидопа 25 |
Стартовая дозировка 62,5 мг (1/4 таблетки) внутрь 2 р/день.
Повышать дозировку возможно каждые 5 дней. Максимальная суточная дозировка подбирается индивидуально.
|
С |
Таблица 3.1 – Препараты для лечения брадикинезии и акинетико-ригидного синдрома при БГ у детей [59].
|
Фармакотерапевтическая группа
|
МНН | Дозировка | Уровень доказательности |
| Противопаркинсонический препарат. Дофаминергическое средство. | Леводопа 250/ Карбидопа 25 |
Стартовая суточная дозировка 0,5-1 мг/кг массы тела внутрь в 2 приема.
Максимальная суточная дозировка подбирается индивидуально.
|
С |
Лечение дистонии при БГ
Таблица 4 – Препараты для лечения дистонии, бруксизма при БГ.
|
Фармакотерапевтическая группа
|
МНН | Способ применения | Уровень доказательности |
| Средства, влияющие на нервно-мышечную передачу | Ботулинический токсин типа А |
Подбор дозировок проводится индивидуально в зависимости от возраста, степени выраженности симптомов и конкретно-выбранного препарата.
Введение – в определенные в ходе осмотра мышцы.
|
В |
| Средства, влияющие на нервно-мышечную передачу | Баклофен | Стартовая дозировка 5 мг внутрь 3 р/день. Подбор дозировок проводится индивидуально в зависимости от степени выраженности симптомов до 120 мг/день. | С |
Таблица 4.1 – Препараты для лечения дистонии, бруксизма при БГ в детском возрасте.
|
Фармакотерапевтическая группа
|
МНН | Способ применения | Уровень доказательности |
| Средства, влияющие на нервно-мышечную передачу | Ботулинический токсин типа А | Подбор дозировок проводится индивидуально в зависимости от возраста, степени выраженности симптомов и конкретно-выбранного препарата. Препарат вводится в мышцы-мишени. | В |
| Средства, влияющие на нервно-мышечную передачу | Баклофен | У детей от 3 до 6 лет – 20-30 мг в сутки, от 6 до 10 лет – 30-60 мг в сутки, перорально. У детей старше 10 лет максимальная суточная доза составляет 2,5 мг/кг массы тела; начальная доза – 1,5-2,0 мг/кг массы тела. Подбор дозы индивидуален. | С |
Таблица 5 – Препараты для лечения миоклонуса при БГ.
|
Фармакотерапевтическая группа
|
МНН | Способ применения | Уровень доказательности |
|
Бензодиазепины
|
Клоназепам |
Стартовая дозировка – 0,5 мг внутрь.
Максимальная суточная дозировка: 1-2 мг.
|
В |
| Противосудорожное средство | Вальпроевая кислота | 750 мг в сутки внутрь в 2-3 приема | С |
| Противосудорожное средство | Леветирацетам | 500-750 мг в сутки внутрь в 2 приема | С |
Таблица 5.1 – Препараты для лечения миоклонуса при БГ в детском возрасте
|
Фармакотерапевтическая группа
|
МНН | Способ применения | Уровень доказательности |
|
Бензодиазепины
|
Клоназепам |
Дети принимают препарат 2-3 раза в сутки, перорально: в возрасте 1-5 лет начальная доза не более 250 мкг/сут;
дети в возрасте 5-12 лет - 500 мкг/сут, перорально. Поддерживающие суточные дозы для детей в возрасте до 1 года - 0.5-1 мг, 1-5 лет - 1-3 мг, 5-12 лет - 3-6 мг. Подбор дозы индивидуален.
|
В |
| Противосудорожное средство | Вальпроевая кислота | Препарат принимают 2 раза в сутки перорально: начальная доза составляет 10-15 мг/кг/сут с повышением дозы по 5мг/кг/сут еженедельно, до эффективной. Средняя суточная доза составляет 25-30 мг/кг/сут. | С |
| Противосудорожное средство | Леветирацетам | Дети принимают препарат дважды в день, перорально. Начальная доза для детей с массой ниже 50 кг: 10 мг/кг два раза в день. Максимальная доза: 30 мг/кг два раза в день. Доза препарата у детей с массой тела 50 кг и выше аналогична взрослым. | С |
Таблица 6 – Препараты для лечения деменции у пациентов с БГ [48-50].
|
Фармакотерапевтическая группа
|
МНН | Способ применения | Уровень доказательности |
| Антихолинэстеразные препараты | Ривастигмин* | Стартовая дозировка – 1,5 мг внутрь. Максимальная суточная дозировка: 6 мг. | В |
Лечение эпилепсии при БГ
Таблица 7 – Препараты для лечения эпилепсии при БГ (предлагаемые препараты, также стабилизируют настроение при болезни Гентингтона) [19,32].
|
Фармакотерапевтическая группа
|
МНН | Способ применения | Уровень доказательности |
| Противосудорожное средство | Вальпроевая кислота | 750 мг в сутки внутрь в 2-3 приема | С |
| Противосудорожное средство | Карбамазепин |
Начальная доза 100-200 мг в сутки внутрь в 2 приема.
При необходимости постепенное повышение дозы до максимальной 1600 мг в сутки
|
С |
| Противосудорожное средство | Ламотриджин |
Начальная доза 25 мг в сутки внутрь.
При необходимости постепенное повышение дозы до максимальной 400 мг в сутки
|
С |
Таблица 7.1 – Препараты для лечения эпилепсии при БГ в детском возрасте.
|
Фармакотерапевтическая группа
|
МНН | Способ применения | Уровень доказательности |
| Противосудорожное средство | Вальпроевая кислота | Дети принимают препарат дважды в день, перорально. Начальная доза для детей с массой ниже 50 кг: 10 мг/кг; Максимальная доза: 30 мг/кг. | С |
| Противосудорожное средство | Карбамазепин | Дети принимают препарат дважды в день, перорально Начальная доза 100 мг. При необходимости постепенное повышение дозы до максимальной 30-35 мг/кг в сутки. | С |
| Противосудорожное средство | Ламотриджин | Дети принимают препарат 1-2 раза в день, перорально. В первую-вторую недели в дозе 0,3мг/кг/сут; на третьей-четвертой неделе по 0,6 мг/кг/сут. Поддерживающие дозы 2-10-15 мг/кг/сут при монотерапии. Доза препарата у детей с старше 13 лет аналогична взрослым | С |
Таблица 8 – Препараты для лечения тревоги, депрессии, апатии при БГ [32].
|
Фармакотерапевтическая группа
|
МНН | Способ применения | Уровень доказательности |
| Селективный ингибитор обратного захвата серотонина | Циталопрам |
Начинать с 10-20 мг в день внутрь.
Максимальная суточная дозировка: 40 мг (у пациентов более старшего возраста – 20 мг)
|
С |
| Селективный ингибитор обратного захвата серотонина | Сертралин |
Стартовая дозировка: 50 мг внутрь.
Максимальная суточная дозировка: 200 мг.
|
С |
| Селективный ингибитор обратного захвата серотонина | Флуоксетин |
20 мг в сутки внутрь с постепенным повышением.
Максимальная суточная дозировка: 60 мг.
|
С |
| Селективный ингибитор обратного захвата серотонина и норадреналина | Венлафаксин |
75 мг в два приема внутрь ежедневно (по 37.5 мг 2 раза в день) с возможным постепенным повышением до 150 мг/сут.
Максимальная суточная дозировка: 225 мг.
|
С |
| Селективный ингибитор обратного захвата серотонина и норадреналина | Дулоксетин |
30 мг в сутки внутрь с повышением до 60 мг/сут.
Максимальная суточная дозировка: 90 мг.
|
С |
| Тетрациклический антиепрессант | Миртазапин* |
Стартовая дозировка: 15 мг внутрь.
Максимальная суточная дозировка: 45 мг.
|
С |
| Антидепрессанты | Миансерин |
Стартовая дозировка 30 мг внутрь.
Возможно постепенное увеличение дозировки до 90 мг в сутки.
|
С |
| Атипичные нейролептики | Оланзапин** |
Стартовая дозировка: 5 мг внутрь.
Максимальная суточная дозировка: 20 мг.
|
С |
Таблица 8.1 – Препараты для лечения тревоги, депрессии, апатии при БГ в детском возрасте.
|
Фармакотерапевтическая группа
|
МНН | Способ применения | Уровень доказательности |
| Селективный ингибитор обратного захвата серотонина | Сертралин | Детям старше 6 лет, перорально. Стартовая дозировка: 25 мг. Максимальная суточная дозировка: 200 мг. | С |
| Селективный ингибитор обратного захвата серотонина | Флуоксетин |
Детям старше 6 лет, перорально. 20 мг в сутки с постепенным повышением.
Максимальная суточная дозировка: 40 мг.
|
С |
Лечение психозов при БГ
Таблица 9 – Препараты для лечения психозов, галлюцинаций при БГ (рекомендуется проводить совместно с психиатром).
|
Фармакотерапевтическая группа
|
МНН | Способ применения | Уровень доказательности |
| Атипичный нейролептик | Оланзапин |
Стартовая дозировка: 5 мг внутрь.
Максимальная суточная дозировка: 20 мг.
|
В |
| Атипичный нейролептик | Рисперидон | Стартовая дозировка: 0,5 мг 1 раз в день внутрь, при отсутствии положительного эффекта, возможно увеличить до 1 мг (кратность приема по 0,5 мг 2 раза в день). Максимальная суточная дозировка: 4–6 мг | В |
| Атипичный нейролептик | Клозапин |
Если еще присутствуют признаки паркинсонизма: акинетический синдром.
Стартовая дозировка 25 мг внутрь в сутки.
Возможно постепенное повышение на 25 в день.
Максимальная суточная доза 300 мг.
|
С |
Таблица 9.1 – Препараты для лечения психозов, галлюцинаций при БГ у детей (рекомендуется проводить совместно с психиатром) [60].
|
Фармакотерапевтическая группа
|
МНН | Способ применения | Уровень доказательности |
| Атипичный нейролептик | Оланзапин |
Стартовая дозировка: 2,5 мг внутрь.
Максимальная суточная дозировка: 10 мг.
|
В |
| Атипичный нейролептик | Рисперидон | Стартовая дозировка: 0,25 мг 1 раз в день внутрь, при отсутствии положительного эффекта, возможно увеличить до 0,5 мг (кратность приема по 0,25 мг 2 раза в день). Максимальная суточная дозировка: 1 мг | В |
| Атипичный нейролептик | Кветиапин | Стартовая дозировка: 25 мг 1 раз в день внутрь, при отсутствии положительного эффекта, возможно увеличить до 50 мг в день. | С |
Лечение раздражительности, импульсивности.
Таблица 10 – Препараты для лечения раздражительности, импульсивности (перед инициацией медикаментозного лечения необходимо провести оценку возможных внешних раздражителей, дать рекомендации родственникам и ухаживающим о необходимости соблюдения распорядка дня, о возможном применении отвлекающих методик для избежания излишней раздражительности пациента).
|
Фармакотерапевтическая группа
|
МНН | Способ применения | Уровень доказательности |
| Селективный ингибиторы обратного захвата серотонина | Циталопрам |
Начинать с 10-20 мг в день внутрь.
Необходимо постепенно повышать дозировку до максимально приближенной или равной суточной дозировке в 40 мг (у пациентов более старшего возраста – 20 мг)
|
С |
| Селективный ингибиторы обратного захвата серотонина | Сертралин | Стартовая дозировка: 50 мг внутрь с постепенным повышением максимально приближенной или равной суточной дозировке в 200 мг | С |
| Селективный ингибитор обратного захвата серотонина | Флуоксетин | 20 мг в сутки внутрь с постепенным повышением максимально приближенной или равной суточной дозировке в 60 мг. | С |
| Атипичный нейролептик | Оланзапин |
Стартовая дозировка: 5 мг внутрь.
Максимальная суточная дозировка: 20 мг
|
В |
| Противосудорожное средство | Вальпроевая кислота | 750 мг в сутки внутрь в 2-3 приема. | С |
| Противосудорожное средство | Карбамазепин | Начальная доза 100-200 мг внутрь в сутки в 2 приема. При необходимости постепенное повышение дозы до максимальной 1600 мг в сутки | С |
| Тетрациклический антидепрессант | Миртазапин* |
Стартовая дозировка: 15 мг внутрь.
Максимальная суточная дозировка: 45 мг.
|
С |
|
Психоаналептики
Антидепрессанты
|
Миансерин |
Стартовая дозировка 30 мг 1 р/день внутрь.
Возможно постепенное увеличение дозировки до 90 мг в сутки.
|
С |
NB! Хотя СИОЗС являются препаратами первой линии при раздражительности для достижения эффективности может потребоваться их использование в максимально рекомендуемой дозе или близкой к ней. Раздражительным пациентам, которым не помогает применение только СИОЗС, может быть полезна комбинированная терапия с миансерином или миртазапином, особенно при наличии нарушений сна. У пациентов с агрессивным поведением рекомендуемым лечением первой линии являются нейролептик. В случае явной агрессии, связанной с депрессией, лечение нейролептиками следует сочетать с седативными антидепрессантами.
Таблица 10.1 – Препараты для лечения раздражительности, импульсивности в детском возрасте.
|
Фармакотерапевтическая группа
|
МНН | Способ применения | Уровень доказательности |
| Селективный ингибитор обратного захвата серотонина | Сертралин | Детям старше 6 лет, перорально. Стартовая дозировка: 25 мг. Максимальная суточная дозировка: 200 мг. | С |
| Селективный ингибитор обратного захвата серотонина | Флуоксетин |
Детям старше 6 лет, перорально. 20 мг в сутки с постепенным повышением.
Максимальная суточная дозировка: 40 мг.
|
С |
| Противосудорожное средство | Карбамазепин | Дети принимают препарат дважды в день, перорально Начальная доза 50-100 мг. При необходимости постепенное повышение дозы до максимальной 10 мг/кг в сутки. | С |
Лечение нарушений сна.
Таблица 11 – Препараты для лечения нарушений сна.
|
Фармакотерапевтическая группа
|
МНН | Способ применения | Уровень доказательности |
| Тетрациклический антидепрессант | Миртазапин* |
Стартовая дозировка: 15 мг внутрь.
Максимальная суточная дозировка: 45 мг
|
С |
|
Психоаналептики
Антидепрессанты
|
Миансерин |
Стартовая дозировка 30 мг внутрь.
Возможно постепенное увеличение дозировки до 90 мг в сутки
|
С |
| Трициклический антидепрессант | Амитриптилин | 12,5-50 мг/сут | С |
Лечение гиперсаливации.
Таблица 12 – Препараты для лечения гиперсаливации.
|
Фармакотерапевтическая группа
|
МНН | Способ применения | Уровень доказательности |
|
Спазмолитик
|
Скополамин
[51-53]
|
20 мг 3 р/день подкожно | С |
| Блокатор М-холинорецепторов | Атропин [51-53] | 300 мкг 4 р/сут под язык | С |
| Трициклический антидепрессант | Амитриптилин | 12,5-50 мг/сут внутрь | С |
| Средства, влияющие на нервно-мышечную передачу | Ботулинический токсин типа А |
Индивидуально в зависимости от степени выраженности симптомов и выбранного препарата.
Препарат вводится в околоушные и поднижнечелюстные слюнные железы.
|
В |
NB! При назначении атропина, скопаламина следует учитывать ятрогенные риски, в частности состояние спутанности сознания, запор, повышенное внутриглазное давление, задержку мочи.
3. Хирургическое вмешательство: на амбулаторном этапе не предусмотрено.
Реабилитационные мероприятия, направленные на улучшение функционирования и качества жизни пациента, страдающего БГ (см. п.3.1. немедикаментозное лечение).
Планирование семьи
Возможно пренатальное тестирование с помощью молекулярно-генетического тестирования и проведение преимплантационного генетического тестирования.
5. Индикаторы эффективности лечения и безопасности методов лечения.
Независимо от возраста начала и начала симптоматической терапии БГ является хроническим, медленно прогрессирующим заболеванием [45]. Средняя продолжительность выживания после клинического начала колеблется от 10 до 20 лет, некоторые пациенты живут от 30 до 40 лет [46].
Необходимо постоянно мониторировать физическое и психологическое состояние пациентов, своевременно направляя на дополнительное обследование и оказание специализированной помощи, а также осуществлять тщательный уход и оказывать психологическую поддержку.
Мониторинг осложнений, являющихся причиной смерти пациентов с БГ:
- пневмония,
- травмы,
- суицид,
- желудочно-кишечные осложнения,
- урологические осложнения,
- сердечно-сосудистые осложнения [47].
Задача терапии заключается в оптимальном контроле симптомов, поддержании качества жизни пациентов, контролю побочных эффектов терапии.
Лечение (стационар)
1. Карта наблюдения пациента, маршрутизация пациента: нет.
2. Немедикаментозное лечение (режим, диета) – см. п. 1. Немедикаментозное лечение на амбулаторном уровне.
3. Медикаментозное лечение – см. п. 2. Медикаментозное лечение на амбулаторном уровне.
5. Дальнейшее ведение: Реабилитационные мероприятия после хирургического лечения неспецифические – в зависимости от вида проведенного вмешательства.
6. Индикаторы эффективности лечения и безопасности методов диагностики и лечения, описанных в протоколе:
Госпитализация
Показания для плановой госпитализации:
Показания для экстренной госпитализации:
Информация
Источники и литература
-
Протоколы заседаний Объединенной комиссии по качеству медицинских услуг МЗ РК, 2024
- 1. Международная классификация болезней 11 пересмотра, сайт Всемирной Организации Здравоохранения URI Базового компонента: http://id.who.int/icd/entity/2132180242. 2. Rawlins MD, Wexler NS, Wexler AR, Tabrizi SJ, Douglas I, Evans SJ, Smeeth L. The prevalence of Huntington's disease. Neuroepidemiology. 2016;46:144–53. 3. Fisher ER, Hayden MR. Multisource ascertainment of Huntington disease in Canada: prevalence and population at risk. Mov Disord. 2014;29:105–14. 4. Baig SS, Strong M, Rosser E, Taverner NV, Glew R, Miedzybrodzka Z, Clarke A, Craufurd D, Quarrell OW, et al. 22 years of predictive testing for Huntington's disease: the experience of the UK Huntington's Prediction Consortium. Eur J Hum Genet. 2016;24:1396–402. 5. Pringsheim T, Wiltshire K, Day L, Dykeman J, Steeves T, Jette N. The incidence and prevalence of Huntington's disease: a systematic review and meta-analysis. Mov Disord. 2012;27:1083–91. 6. Sipilä JO, Hietala M, Siitonen A, Päivärinta M, Majamaa K. Epidemiology of Huntington's disease in Finland. Parkinsonism Relat Disord. 2015;21:46–9. Xu M, Wu ZY. Huntington disease in Asia. Chin Med J (Engl). 2015;128:1815–9. 7. Caron NS, Wright GEB, Hayden MR. Huntington Disease. 1998 Oct 23 [Updated 2020 Jun 11]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1305. 8. Kwa L, Larson D, Yeh C, Bega D. Influence of Age of Onset on Huntington's Disease Phenotype. Tremor Other Hyperkinet Mov (N Y). 2020 Jul 9;10:21. doi: 10.5334/tohm.536. PMID: 32775035; PMCID: PMC7394225. 9. Ranganathan M, Kostyk SK, Allain DC, Race JA, Daley AM. Age of onset and behavioral manifestations in Huntington's disease: An Enroll-HD cohort analysis. Clin Genet. 2021 Jan;99(1):133-142. doi: 10.1111/cge.13857. Epub 2020 Oct 16. PMID: 33020896. 10. Huntington Study Group. Unified Huntington’s Disease Rating Scale: Reliability and-Consis tenc y Movement Disorders Vol. II, No. 2, 1996, pp. 136142. 11. Jacobs M, Hart EP, van Zwet EW, Bentivoglio AR, Burgunder JM, Craufurd D, Reilmann R, Saft C, Roos RA; REGISTRY investigators of the European Huntington’s Disease Network. Progression of motor subtypes in Huntington's disease: a 6-year follow-up study. J Neurol. 2016 Oct;263(10):2080-5. doi: 10.1007/s00415-016-8233-x. Epub 2016 Jul 19. PMID: 27435968; PMCID: PMC5037142. 12. McCusker EA, Gunn DG, Epping EA, Loy CT, Radford K, Griffith J, Mills JA, Long JD, Paulsen JS; PREDICT-HD Investigators of the Huntington Study Group. Unawareness of motor phenoconversion in Huntington disease. Neurology. 2013 Sep 24;81(13):1141-7. doi: 10.1212/WNL.0b013e3182a55f05. Epub 2013 Aug 21. PMID: 23966256; PMCID: PMC3795599. 13. McCusker E, Loy CT. The many facets of unawareness in Huntington disease. Tremor Other Hyperkinet Mov (N Y). 2014 Nov 12;4:257. doi: 10.7916/D8FJ2FD3. PMID: 25411649; PMCID: PMC4231168. 14. Squitieri F, Frati L, Ciarmiello A, Lastoria S, Quarrell O. Juvenile Huntington’s disease: Does a dosage-effect pathogenic mechanism differ from the classical adult disease? Mechanisms of Ageing and Development 2006;127:208–12. 15. Telenius H, Kremer HP, Theilmann J, et al: Molecular analysis of juvenile Huntington disease: The major influence on (CAG)n repeat length is the sex of the affected parent. Hum Mol Genet 1993;2:1535-1540. 16. Landau ME, Cannard KR: EEG characteristic in juvenile Huntington’s disease: A case report and review of the literature. Epileptic Disord 2003;53:1–4. 17. Kringlen G, Kinsley L, Aufox S, Rouleau G, Bega D. The Impact of Family History on the Clinical Features of Huntington's Disease. J Huntingtons Dis. 2017;6(4):327-335. doi: 10.3233/JHD-170256. PMID: 28984613. 18. Semaka A, Kay C, Doty C, Collins JA, Bijlsma EK, Richards F, Goldberg YP, Hayden MR. CAG size-specific risk estimates for intermediate allele repeat instability in Huntington disease. J Med Genet. 2013a;50:696–703. 19. Oksana Suchowersky. Huntington disease: Clinical features and diagnosis – UpToDate Literature review: Apr 2023. 20. Nance MA, Myers RH: Juvenile onset Huntington’s disease—Clinical and research perspectives. Ment Retard Dev Disabil Res Rev 2001;7: 153–157. 21. Nance MA, Mathis-Hagen V, Breningstall G, et al: Analysis of very large trinucleotide repeat in patient with juvenile Huntington’s disease. Neurology 1999;52:392–394. 22. Tabrizi SJ, Scahill RI, Owen G, Durr A, Leavitt BR, Roos RA, Borowsky B, Landwehrmeyer B, Frost C, Johnson H, Craufurd D, Reilmann R, Stout JC, Langbehn DR, et al. Predictors of phenotypic progression and disease onset in premanifest and early-stage Huntington's disease in the TRACK-HD study: analysis of 36-month observational data. Lancet Neurol. 2013;12:637–49. 23. Gambardella A, Muglia M, Labate A, Magariello A, Gabriele L, Mazzei R, et al. Juvenile Huntington’s disease presenting as progressive myoclonic epilepsy. Neurology 2001;57:708-11. 24. Saft C, Andrich JE, Müller T, Becker J, Jackowski J. Oral and dental health in Huntington's disease - an observational study. BMC Neurol. 2013 Sep 3;13:114. doi: 10.1186/1471-2377-13-114. PMID: 24138900; PMCID: PMC3766132. 25. Vicars BG, Liu AB, Holt S, Jayadev S, Bird T, Yang CC. High Frequency of Concomitant Bladder, Bowel, and Sexual Symptoms in Huntington's Disease: A Self-Reported Questionnaire Study. J Pers Med. 2021 Jul 25;11(8):714. doi: 10.3390/jpm11080714. PMID: 34442358; PMCID: PMC8401810. 26. Jankovic J. et al. Principles and Practice of Movement Disorders. 3rd Edition. Elsevier. 2022. 627 pp. 27. Sciacca S, Favellato M, Madonna M, Metro D, Marano M, Squitieri F. Early enteric neuron dysfunction in mouse and human Huntington disease. Parkinsonism Relat Disord. (2017) 34:73–4. doi: 10.1016/j.parkreldis.2016.10.017) 28. Trejo A, Boll M-C, Alonso ME, Ochoa A, Velásquez L. Use of oral nutritional supplements in patients with Huntington's disease. Nutrition. (2005) 21:889–94. doi: 10.1016/j.nut.2004.12.012. 29. Trejo A, Boll M-C, Alonso ME, Ochoa A, Velásquez L. Use of oral nutritional supplements in patients with Huntington's disease. Nutrition. (2005) 21:889–94. doi: 10.1016/j.nut.2004.12.012 30. Rivadeneyra J, Cubo E, Polo CG, Mariscal N, Calvo S, Mateos A, et al. J14 mediterranean diet and nutritional composition of patients with Huntington's disease. Spanish multicenter study of the European group for Huntington's disease. J Neurol Neurosurg Psychiatry. (2014) 85(Suppl. 1):A69–70. doi: 10.1136/jnnp-2014-309032.197. 31. Cubo E, Rivadeneyra J, Mariscal N, Armesto D, Mateos A, Camara R. The relationship between dietary intake, nutrition status and Huntington's disease severity (P1.050). Neurology. (2014) 4:78–85. 32. Patrick, C. and Ritchie, S. (2020), Managing the symptoms of Huntington's disease. Prescriber, 31: 23-27. https://doi.org/10.1002/psb.1824. 33. Heemskerk AW, Roos RA. Dysphagia in Huntington's disease: a review. Dysphagia. 2011 Mar;26(1):62-6. doi: 10.1007/s00455-010-9302-4. Epub 2010 Sep 14. PMID: 20838817. 34. Monaco AD, Nuzzi A, Parente A, Lavermicocca V, Chiarelli T, Tommaso MD, et al. I03 swallowing function in the early, middle and late stages of Huntington's disease. J Neurol Neurosurg Psychiatry. (2014) 85(Suppl. 1):A58. doi: 10.1136/jnnp-2014-309032.165. 35. Wheelock V. The motor disorder. In: A Physician's Guide to the Management of Huntington's Disease, 3rd ed, Nance M, Paulsen JS, Rosenblatt A, Wheelock V (Eds), Huntington's Disease Society of America, 2011. p.39. 36. Fiedorowicz JG, Mills JA, Ruggle A, Langbehn D, Paulsen JS PREDICT-HD Investigators of the Huntington Study Group. Suicidal behavior in prodromal Huntington disease. Neurodegener Dis. (2011) 8:483–90. doi: 10.1159/000327754. 37. Wesson M, Boileau NR, Perlmutter JS, Paulsen JS, Barton SK, McCormack MK, et al. Suicidal ideation assessment in individuals with premanifest and manifest Huntington disease. J Huntingtons Dis. (2018) 7:239–49. doi: 10.3233/JHD-180299. 38. Quinn L, Kegelmeyer D, Kloos A, Rao AK, Busse M, Fritz NE. Clinical recommendations to guide physical therapy practice for Huntington disease. Neurology. 2020 Feb 4;94(5):217-228. doi: 10.1212/WNL.0000000000008887. Epub 2020 Jan 6. PMID: 31907286; PMCID: PMC7080285. 39. Quinn L et al. Clinical recommendations to guide physical therapy practice for Huntington disease. Neurology. 2020;94(5):217. Epub 2020 Jan 6. 40. Lewis D, Fiske J, Dougall A. Access to special care dentistry, part 7. Special care dentistry services: seamless care for people in their middle years–part 1. Br Dent J. (2008) 205:305–17. doi: 10.1038/sj.bdj.2008.803. 41. Manley G, Lane H, Carlsson A, Ahlborg B, Mårtensson Å, Nilsson MB, et al. Guideline for oral healthcare of adults with Huntington's disease. Neurodegener Dis Manag. (2012) 2:55–65. doi: 10.2217/nmt.11.68. 42. Boyle CA, Frölander C, Manley G. Providing dental care for patients with Huntington's disease. Dent Update. (2008) 35:333–6. doi: 10.12968/denu.2008.35.5.333. 43. Cangemi CF, Miller RJ. Huntington's disease: review and anesthetic case management. Anesth Prog. (1998) 45:150–3. 44. Tarolli CG, Chesire AM, Biglan KM. Palliative Care in Huntington Disease: Personal Reflections and a Review of the Literature. Tremor Other Hyperkinet Mov (N Y). 2017 Apr 11;7:454. doi: 10.7916/D88057C7. PMID: 28428907; PMCID: PMC5395679. 45. Dorsey E.R. et al. Natural history of Huntington disease. JAMA Neurol. 2013 Dec;70(12):1520-30. 46. Rosenblatt A. Overview and principles of treatment. In: A Physician's Guide to the Management of Huntington's Disease, 3rd ed, Huntington's Disease Society of America, 2011. p.5. 47. Solberg OK, Filkuková P, Frich JC, Feragen KJB. Age at Death and Causes of Death in Patients with Huntington Disease in Norway in 1986-2015. J Huntingtons Dis. 2018;7(1):77-86. doi: 10.3233/JHD-170270. PMID: 29480207; PMCID: PMC5870025. 48. de Tommaso M, Difruscolo O, Sciruicchio V, Specchio N, Livrea P. Two years' follow-up of rivastigmine treatment in Huntington disease. Clin Neuropharmacol. 2007 Jan-Feb;30(1):43-6. doi: 10.1097/01.wnf.0000240945.44370.f0. PMID: 17272969. 49. de Tommaso M, Specchio N, Sciruicchio V, Difruscolo O, Specchio LM. Effects of rivastigmine on motor and cognitive impairment in Huntington's disease. Mov Disord. 2004 Dec;19(12):1516-8. doi: 10.1002/mds.20235. PMID: 15390067. 50. Puneet Kumar, Anil Kumar, Protective effect of rivastigmine against 3-nitropropionic acid-induced Huntington's disease like symptoms: Possible behavioural, biochemical and cellular alterations, European Journal of Pharmacology, Volume 615, Issues 1–3, 2009, Pages 91-101, ISSN 0014-2999, https://doi.org/10.1016/j.ejphar.2009.04.058. 51. Saft C, Burgunder JM, Dose M, Jung HH, Katzenschlager R, Priller J, Nguyen HP, Reetz K, Reilmann R, Seppi K, Landwehrmeyer GB. Symptomatic treatment options for Huntington's disease (guidelines of the German Neurological Society). Neurol Res Pract. 2023 Nov 16;5(1):61. doi: 10.1186/s42466-023-00285-1. PMID: 37968732; PMCID: PMC10652593. 52. Bachoud-Lévi AC, Ferreira J, Massart R, Youssov K, Rosser A, Busse M, Craufurd D, Reilmann R, De Michele G, Rae D, Squitieri F, Seppi K, Perrine C, Scherer-Gagou C, Audrey O, Verny C, Burgunder JM. International Guidelines for the Treatment of Huntington's Disease. Front Neurol. 2019 Jul 3;10:710. doi: 10.3389/fneur.2019.00710. PMID: 31333565; PMCID: PMC6618900. 53. Huntington’s Disease Association. Care in advanced Huntington’s disease. 2019. 54. Tetrabenazine Therapy of Pediatric Hyperkinetic Movement Disorders Samay Jain, MD,1* Paul E. Greene, MD,2 and Steven J. Frucht Mov Disord. 2006 Nov;21(11):1966-72. doi: 10.1002/mds.21063. 55. Coffey B, Jankovic J, Claassen DO, Jimenez-Shahed J, Gertz BJ, Garofalo EA, Stamler DA, Wieman M, Savola JM, Gordon MF, Alexander JK, Barkay H, Harary E. Efficacy and Safety of Fixed-Dose Deutetrabenazine in Children and Adolescents for Tics Associated With Tourette Syndrome: A Randomized Clinical Trial. JAMA Netw Open. 2021 Oct 1;4(10):e2129397 56. Jankovic J, Coffey B, Claassen DO, Jimenez-Shahed J, Gertz BJ, Garofalo EA, Stamler DA, Wieman M, Savola JM, Harary E, Alexander J, Barkay H, Gordon MF. Safety and Efficacy of Long-Term Deutetrabenazine Use in Children and Adolescents with Tics Associated with Tourette Syndrome: An Open-Label Extension Study. Mov Disord Clin Pract. 2023 Aug 24;10(9):1388-1398. 57. https://www.drugs.com/dosage/risperidone.html 58. https://www.drugs.com/dosage/haloperidol.html #Usual_Pediatric_Dose_for_Tourette_s_Syndrome 59. Hoshino K, Hayashi M, Ishizaki A, Kimura K, Kubota M, Nezu A, Yasuhara A. Very-Low-Dose Levodopa Therapy for Pediatric Neurological Disorders: A Preliminary Questionnaire in Japan. Front Pediatr. 2021 Mar 4;9:569594. doi: 10.3389/fped.2021.569594. PMID: 33748036; PMCID: PMC7970027. 60. Stevens JR, Prince JB, Prager LM, Stern TA. Psychotic disorders in children and adolescents: a primer on contemporary evaluation and management. Prim Care Companion CNS Disord. 2014;16(2):PCC.13f01514. doi: 10.4088/PCC.13f01514. Epub 2014 Mar 13. PMID: 25133052; PMCID: PMC4116281.
Информация
Список разработчиков протокола с указание квалификационных данных:
Протокол был разработан по запросу Общественного Объединения «Общество пациентов с редкими генетическими заболеваниями «Сенім».
Конфликт интересов: отсутствует.
Рецензенты:
Указания условий пересмотра протокола: Пересмотр не реже 1 раза в 5 лет и не чаще 1 раза в 3 года при наличии новых методов диагностики и лечения с уровнем доказательности.
Total Functional Capacity – Оценка общей функциональной способности
Функционирование на работе
Финансы
Домашние дела
Ежедневная активность
Уровень ухода
Итого:
Прикреплённые файлы
Внимание!
- Занимаясь самолечением, вы можете нанести непоправимый вред своему здоровью.
- Информация, размещенная на сайте MedElement и в мобильных приложениях "MedElement (МедЭлемент)", "Lekar Pro", "Dariger Pro", "Заболевания: справочник терапевта", не может и не должна заменять очную консультацию врача. Обязательно обращайтесь в медицинские учреждения при наличии каких-либо заболеваний или беспокоящих вас симптомов.
- Выбор лекарственных средств и их дозировки, должен быть оговорен со специалистом. Только врач может назначить нужное лекарство и его дозировку с учетом заболевания и состояния организма больного.
- Сайт MedElement и мобильные приложения "MedElement (МедЭлемент)", "Lekar Pro", "Dariger Pro", "Заболевания: справочник терапевта" являются исключительно информационно-справочными ресурсами. Информация, размещенная на данном сайте, не должна использоваться для самовольного изменения предписаний врача.
- Редакция MedElement не несет ответственности за какой-либо ущерб здоровью или материальный ущерб, возникший в результате использования данного сайта.