Эпилепсия и эпилептические синдромы у детей
![]() Версия: Клинические протоколы КР 2022 (Кыргызстан)
Версия: Клинические протоколы КР 2022 (Кыргызстан)
Общая информация
Краткое описание
Приложение 9
к приказу МЗ КР № 335
от “16” марта 2022 г.
МИНИСТЕРСТВО ЗДРАВООХРАНЕНИЯ КЫРГЫЗСКОЙ РЕСПУБЛИКИ
НАЦИОНАЛЬНЫЙ ЦЕНТР ОХРАНЫ МАТЕРИНСТВА ИДЕТСТВА
КЫРГЫЗСКАЯ ГОСУДАРСТВЕННАЯ МЕДИЦИНСКАЯ АКАДЕМИЯ
КЫРГЫЗСКО-РОССИЙСКИЙ СЛАВЯНСКИЙ УНИВЕРСИТЕТ
КЛИНИЧЕСКОЕ РУКОВОДСТВО
ПО ДИАГНОСТИКЕ И ЛЕЧЕНИЮ ЭПИЛЕПСИИ, ЭПИЛЕПТИЧЕСКИХ СИНДРОМОВ У ДЕТЕЙ
КЛИНИЧЕСКАЯ ПРОБЛЕМА
Эпилепсия, эпилептические синдромы у детей и подростков (G40.0-G40.9)
НАЗВАНИЕ ДОКУМЕНТА
Клиническое руководство по диагностике и лечению эпилепсии, эпилептических синдромов у детей
ЭТАПЫ ОКАЗАНИЯ ПОМОЩИ
Первичная медицинская помощь
Специализированная помощь на первичном уровне/ПМСП
Специализированная помощь на вторичном уровне.
Специализированная помощь на третичном уровне.
ДАТА СОЗДАНИЯ: 2020г.
Пересмотр руководства планируется через 2 года с момента утверждения.
ЦЕЛЬ СОЗДАНИЯ РУКОВОДСТВА
предоставление врачам ГСВ, ЦСМ, ТБ, ООБ и учреждениям третичного уровня рекомендаций по диагностике, лечению и диспансерного наблюдения детей и подростков с эпилептическими приступами (эпилепсией и эпилептических синдромов).
ЦЕЛЕВЫЕ ГРУППЫ
Детские неврологи, детские психиатры, педиатры, врачи центров семейной медицины, социальные работники, детские психологи.
Клиническое руководство применимо для детей и подростков.
Основой целью
разработанного руководства является предоставление специалистам системы здраво- охранения удобных и выполнимых рекомендаций по диагностике, регистрации, перенаправлению и лечению детей и подростков с эпилепсией и эпилептическими синдромами.
Целевые группы пользователей:
Детские неврологи Детские психиатры
Врачи центров семейной медицины.
ВВЕДЕНИЕ:
Актуальность проблемы.
Эпилепсия - одно из самых частых заболеваний нервной системы у детей и подростков, которое занимает третье место в структуре болезней нервной системы. В 60% случаев дебют эпилепсии приходится на детский возраст. Несвоевременное купирование приступов может иметь глубокие и далеко идущие последствия в виде нарушения когнитивных функций, поведения, изменения характера и формирования патологической личности. Эпилептические припадки могут представлять и серьезные угрозу для жизни. Смерть во время припадка может возникать из-за сопутствующих вегетативных нарушений, что 3,2% случаев, или в результате несчастного случая – травмы или утопления. Травма является наиболее частым последствием эпилептического припадка и наблюдается в 30%, а утопление – в 19% случаев. Как и любой припадок мозгового происхождения, эпилептический припадок оказывает огромное психологическое воздействие на окружающих, даже на медицинский персонал. Это приводит к тому, что доступ к дошкольным и школьным учреждениям у этих детей ограничен. Диагноз эпилепсии часто бывает субъективен и основывается лишь на одних анамнестических сведениях, чего совершенно недостаточно. На современном уровне он должен быть верифицирован. Необходима система видео-ЭЭГ-мониторинга, позволяющая детально проанализировать развитие припадка, установить локальный характер и тип приступов, степень нарушения сознания синхронно с электроэнцефалографическими изменениями. Только такой подход в диагностике позволит детально описать и выделить формы эпилепсии, а также обосновать назначение соответствующих антиконвульсантов.
Помощь детям с эпилептическими припадками должна быть комплексной и не может ограничиваться только биологической терапией. В связи с этим, в стандарты оказания помощи этим пациентам необходимо включение психосоциальной терапии и психосоциальной реабилитации, использование психотерапии, клинико-социальных мероприятий и различных организационных форм помощи. Это соответствует современным положениям о мультидисциплинарном подходе в реабилитации пациентов, с психическими расстройствами при обязательном межведомственном взаимодействии.
ЭПИЛЕПСИЯ, ЭПИЛЕПТИЧЕСКИЕ СИНДРОМЫ У ДЕТЕЙ
Код по МКБ 10: Таблица №3
|
Коды МКБ-10
|
|
|
G40
|
Эпилепсия
|
|
G40.0
|
Локализованная (фокальная) (парциальная) идиопатическая эпилепсия и эпилептические синдромы с судорожными приступами с фокальным началом
|
|
G40.1
|
Локализованная (фокальная) (парциальная) симптоматическая эпилепсия и эпилептические синдромы с простыми парциальными приступами
|
|
G40.2
|
Локализованная (фокальная) (парциальная) симптоматическая эпилепсия и эпилептические синдромы с комплексными парциальными судорожными приступами
|
|
G40.3
|
Генерализованная идиопатическая эпилепсия и эпилептические синдромы
|
|
G40.4
|
Другие виды генерализованной эпилепсии и эпилептических синдромов
|
|
G40.5
|
Особые эпилептические синдромы
|
|
G40.6
|
Приступы grand mal неуточненные (с малыми припадками [petit mal] или без них)
|
|
G40.7
|
Малые припадки [petit mal] неуточненные без припадков grand mal
|
|
G40.8
|
Другие уточненные формы эпилепсии
|
|
G40.9
|
Эпилепсия неуточненная
|
|
G 41
|
Эпилептический статус
|
|
G 41.0
|
Эпилептический статус grand mal (судорожных припадков)
|
|
G 41.1
|
Эпилептический статус petit mal (малых припадков)
|
|
G 41.2
|
Сложный парциальный эпилептический статус
|
|
G 41.8
|
Другой уточненный эпилептический статус
|
|
G 41.9
|
Эпилептический статус неуточненный
|
ОПРЕДЕЛЕНИЕ
Согласно консенсусу ILAE и Международного бюро по эпилепсии (International Bureau for Epilepsy, IBE), эпилепсия представляет собой болезнь, включающую различные расстройства и состояния. Решено отказаться от термина «расстройство» или «группа расстройств», поскольку данный термин указывает на раз- личной продолжительности функциональные нарушения, в то время как термин «болезнь» предполагает более длительное нарушение функции. До этого под эпилепсией понимали расстройство головного мозга, характеризующееся стойкой предрасположенностью к эпилептическим приступам, то есть наличие двух неспровоцированных эпилептических приступов с интервалом более 24 ч. В декабре 2013 г. была принята исполнительным комитетом ILAE, а в начале 2014 г. опубликована официальная позиция ILAE в отношении рабочего определения эпилепсии для клинической диагностики. Согласно данному определению; «Эпилепсией считают заболевание головного мозга, отвечающее следующим критериям: 1) не менее двух неспровоцированных (или рефлекторных) эпилептических приступов с интервалом более 24 ч; 2) один неспровоцированный (или рефлекторный) приступ и вероятность повторения приступов, близкая к общему риску рецидива (≥60%) после двух спонтанных приступов, в последующие 10 лет; 3) диагноз эпилептического синдрома».
Эпилептический приступ - это преходящие клинические проявления патологи- чески избыточной или синхронной нейронной активности головного мозга. Приступ, связанный с воздействием какого-либо преходящего фактора на нормальный голов- ной мозг, временно снижающий судорожный порог, не относится к эпилепсии. Синонимами термина «спровоцированный приступ» являются «реактивный приступ», или «острый симптоматический приступ».
Следует различать причину и провоцирующие факторы, так как некоторые состояния (причины) могут создавать длительную предпосылку к эпилептическим приступам. Например, опухоль головного мозга в отличие от инсульта может быть причиной повторных судорожных приступов. Рецидивирующие рефлекторные эпилептические приступы (например, в ответ на вспышки света) являются спровоцированными приступами, которые относят к эпилепсии. Однако судорожный приступ после сотрясения головного мозга на фоне лихорадки (примеры спровоцированных приступов) согласно позиции ILAE не относятся к эпилепсии. Термин «неспровоцированный» предполагает отсутствие временного или обратимого фактора, снижающего су- дорожный порог и вызывающего приступ в указанный момент времени.
В определение эпилепсии добавлен такой критерий, как риск рецидива. Если риск рецидива высок, то даже после одного неспровоцированного приступа следует придерживаться тактики ведения больного с эпилепсией. После двух неспровоцированных приступов он составляет примерно 60-90%. У детей с эпилептиформными изменениями на ЭЭГ риск рецидива в течение 2-3 лет после первого судорожного при- ступа составляет до 56-71%.
Термин «излечение» не был одобрен, поскольку он указывает на то, что риск возобновления эпилептических приступов не выше, чем у здоровых людей, но у пациентов с эпилепсией в анамнезе такой низкий уровень риска никогда не достигается.
Термин «ремиссия» также не был одобрен, поскольку он не указывает на отсутствие болезни и недостаточно ясен для населения. Рабочая группа ILAE одобрила к использованию термин «разрешение». Данный термин свидетельствует, что эпилепсии у пациента уже нет, но вместе с тем нельзя с уверенностью исключить появление приступов в будущем. В качестве критериев разрешения эпилепсии, ILAE рекомендует использовать достижение определенного возраста у пациентов с зависящим от возраста эпилептическим синдромом, либо отсутствие эпилептических приступов в течение 10 лет. У пациентов, не использовавших антиэпилептические препараты (АЭП) не менее 5 лет.
Классификация
Классификации эпилепсии
В современной эпилептологии известно несколько классификаций эпилептических приступов иэпилепсии, а также этапы появления ключевых дефиниций:
• 1961 год. W.G. Lennox. Попытка создания классификации приступов и этиологической классификации эпилепсии.
• 1970 год. H. Gastaut. Фактически первая, принятая ILAE, классификация эпилептических приступов и форм эпилепсии.
• 1981 год. Комиссия ILAE – классификация эпилептических приступов. (Продержалась 36 лет!)
• 1989 год. Комиссия ILAE – классификация эпилептических синдромов. (Ныне действующая!)
• 2001 год. W.T. Blume. «Глоссарий иктальной терминологии».
• 2001 год. J. Engel. Диагностическая схема при эпилепсии.
• 2011 год. Комиссия ILAE – этиологическая классификация эпилепсий.
• 2017 год. Комиссия ILAE – новая классификация эпилептических приступов!
В 1981 г. ILAE представила Стандартизованную классификацию и терминологию эпилептических приступов, которая получила наибольшее распространение в профессиональном сообществе (см. табл. 1).
В 1985 г. ILAE представила Классификацию эпилепсий и эпилептических синдромов, за которой вскоре последовал ее пересмотренный вариант, ратифицированный Генеральной Ассамблеей ILAE в 1989 г.. Классификация эпилепсий и эпилептических синдромов ILAE 1989 г. широко используется во всем мире, оказывая большое влияние на ведение пациентов с эпилепсий и научные исследования (см. табл. 2).
В течение времени, прошедшего после утверждения Международной классификации эпилептических приступов 1981 г., периодически рассматривались предложения по внесению изменений, ряд из которых был принят. Так, в 2010 г. «криптогенные» формы эпилепсии было предложено заменить на «вероятно симптоматические», «парциальные» (приступы и формы эпилепсии) – на «фокальные», а слово «судороги» – на «приступы». Также было исключено деление парциальных(фокальных) приступов на простые и сложные (в зависимости от нарушения сознания).
Таблица №4.
Классификация эпилептических приступов Международной Противоэпилептической Лиги 1981 г.
|
I.
|
Парциальные (фокальные, локальные) припадки
|
|||
|
А.
|
Простые парциальные припадки (без нарушения сознания)
|
|||
|
1.
|
Моторные припадки
|
|||
|
а) фокальные моторные без марша
|
||||
|
б) фокальные моторные с маршем (джексоновские)
|
||||
|
в) адверсивные
|
||||
|
г) постуральные
|
||||
|
д) фонаторные (вокализация или остановка речи)
|
||||
|
2.
|
Соматосенсорные припадки или припадки со специальными сенсорными симптомами (простые галлюцинации, например, вспышки пламени, звон)
|
|||
|
а) соматосенсорные
|
||||
|
б) зрительные
|
||||
|
в) слуховые
|
||||
|
г) обонятельные
|
||||
|
д) вкусовые
|
||||
|
е) с головокружением
|
||||
|
3.
|
Припадки с вегетативно-висцеральными проявлениями (сопровождаются эпигастральными ощущениями, потливостью, покраснением лица, сужением и расширением зрачков)
|
|||
|
4.
|
Приступы с нарушением психических функций (изменения высшей нервной деятельности); редко бывают без нарушения сознания, чаще проявляются как сложные парциальные припадки
|
|||
|
а) дисфазические
|
||||
|
б) дисмнестические (например, ощущение «уже виденного»)
|
||||
|
в) с нарушением мышления (например, мечтательные состояния, нарушение чувства времени)
|
||||
|
г) аффективные (страх, злоба и др.)
|
||||
|
д) иллюзорные (например, макропсия)
|
||||
|
е) сложные галлюцинаторные (например, музыка, сцены)
|
||||
|
Б.
|
Сложные парциальные припадки (с нарушением сознания, могут иногда начинаться с простой симптоматики)
|
|||
|
|
|
1.
|
Простой парциальный припадок с последующим нарушением сознания
|
|
|
а) начинается с простого парциального припадка (А.1 -А.4) с последующим нарушением сознания
|
||||
|
б) с автоматизмами
|
||||
|
2.
|
Начинается с нарушения сознания
|
|||
|
|
|
|
|
|
|
|
|
|
а) только с нарушением сознания
|
|
|
б) с двигательными автоматизмами
|
||||
|
В.
|
Парциальные припадки с вторичной генерализацией (могут быть генерализованными тонико-клоническими, тоническими, клоническими)
|
|||
|
1.
|
Простые парциальные припадки (А), переходящие в генерализованные
|
|||
|
2.
|
Сложные парциальные припадки (Б), переходящие в генерализованные
|
|||
|
3.
|
Простые парциальные припадки, переходящие в сложные, а затем в генерализованные
|
|||
|
II.
|
Генерализованные припадки (судорожные и бессудорожные)
|
|||
|
А.
|
Абсансы
|
|||
|
1.
|
Типичные абсансы
|
|||
|
а) только с нарушением сознания
|
||||
|
б) со слабо выраженным клоническим компонентом
|
||||
|
в) с атоническим компонентом
|
||||
|
г) с тоническим компонентом
|
||||
|
д) с автоматизмами
|
||||
|
е) с вегетативным компонентом
|
||||
|
2.
|
Атипичные абсансы
|
|||
|
а) изменения тонуса более выражены, чем при типичных абсансах
|
||||
|
б) начало и (или) прекращение припадков происходит не внезапно, а постепенно
|
||||
|
Б.
|
Миоклонические припадки (единичные или множественные миоклонические судороги)
|
|||
|
В.
|
Клонические припадки
|
|||
|
Г.
|
Тонические припадки
|
|||
|
Д.
|
Тонико-клонические припадки
|
|||
|
Е.
|
Атонические (астатические) припадки
|
|||
|
III.
|
Неклассифицированные эпилептические припадки
|
|||
|
|
|
Припадки, которые нельзя включить ни в одну из вышеописанных групп из-за отсутствия необходимой информации, а также некоторые неонатальные припадки, например, ритмические движения глаз, жевательные, плавательные движения
|
||
|
|
|
|
|
|
Международная классификация эпилепсий и эпилептических синдромов ILAE, 1989.
|
1.
|
Локализационно-обусловленные формы (фокальные, локальные, парциальные) эпилепсий и эпилептических синдромов
|
||
|
1.1.
|
Идиопатические (с возраст-зависимым началом)
|
||
|
1.1.1.
|
Доброкачественная эпилепсия детского возраста с центрально-темпоральными спайками
|
||
|
1.1.2.
|
Детская эпилепсия с затылочными пароксизмами на ЭЭГ
|
||
|
1.1.3.
|
Первичная эпилепсия чтения
|
||
|
1.2.
|
Симптоматические
|
||
|
1.2.1.
|
Хроническая прогредиентная парциальная эпилепсия детского возраста (синдром Кожевникова)
|
||
|
1.2.2.
|
Синдромы, характеризующиеся припадками, вызываемыми специфическими провоцирующими факторами (включают парциальные припадки вследствие внезапного возбуждения или эмоционального воздействия)
|
||
|
1.2.3.
|
Височно-долевая эпилепсия
|
||
|
1.2.4.
|
Лобно-долевая эпилепсия
|
||
|
1.2.5.
|
Теменно-долевая эпилепсия
|
||
|
1.2.6.
|
Затылочно-долевая эпилепсия
|
||
|
1.3
|
Криптогенные
|
||
|
2.
|
Эпилепсия и синдромы с генерализованными приступами
|
||
|
2.1
|
Идиопатические (с возраст-зависимым началом)
|
||
|
2.1.1.
|
Доброкачественные семейные судороги новорожденных
|
||
|
2.1.2.
|
Доброкачественные идиопатические судороги новорожденных
|
||
|
2.1.3.
|
Доброкачественная младенческая миоклоническая эпилепсия
|
||
|
2.1.4.
|
Детская абсанс-эпилепсия (пикнолепсия)
|
||
|
2.1.5.
|
Ювенильная абсанс-эпилепсия
|
||
|
2.1.6.
|
Ювенильная миоклоническая эпилепсия (импульсивный малый припадок)
|
||
|
2.1.7.
|
Эпилепсия с генерализованными тонико-клоническими судорогами при пробуждении
|
||
|
|
.
|
2.1.8.
|
Другие генерализованные эпилепсии (не указанные выше)
|
|
2.1.9.
|
Эпилепсии со специфическими провоцирующими факторами (рефлекторные припадки, стартл-эпилепсия)
|
||
|
2.2.
|
Криптогенные или симптоматические
|
||
|
2.2.1.
|
Синдром Уэста (инфантильные спазмы)
|
||
|
2.2.2.
|
Синдром Леннокса-Гасто
|
||
|
|
|
2.2.3.
|
Эпилепсия с миоклоническими абсансами
|
|
2.2.4.
|
Эпилепсия с миоклонически-астатическими припадками
|
||
|
2.3.
|
Симптоматические
|
||
|
2.3.1.
|
Неспецифической этиологии
|
||
|
2.3.1.1. Ранняя миоклоническая энцефалопатия
|
|||
|
2.3.1.2. Ранняя младенческая эпилептическая энцефалопатия с участками подавления биоэлектрической активности на ЭЭГ
|
|||
|
2.3.1.3. Другие симптоматические генерализованные формы эпилепсии, не указанные выше
|
|||
|
2.3.2.
|
Специфические синдромы (включают заболевания, при которых припадки являются ранним и основным проявлением болезни)
|
||
|
3.
|
Эпилепсия и синдромы, неопределенные относительно того, являются ли они фокальными или генерализованными
|
||
|
3.1.
|
С генерализованными и фокальными припадками
|
||
|
3.1.1.
|
Судороги новорожденных
|
||
|
3.1.2.
|
Тяжелая миоклоническая эпилепсия раннего детского возраста
|
||
|
3.1.3.
|
Эпилепсия с непрерывными пик-волнами на ЭЭГ в медленной фазе сна
|
||
|
3.1.4.
|
Приобретенная эпилептическая афазия (синдром Ландау-Клеффнера)
|
||
|
3.1.5.
|
Другие формы, не указанные выше
|
||
|
3.2.
|
Без определенных генерализованных и фокальных признаков
|
||
|
4.
|
Специальные синдромы
|
||
|
4.1.
|
Припадки, связанные с определенной ситуацией
|
||
|
4.1.1.
|
Фебрильные судороги
|
||
|
4.1.2.
|
Изолированные единичные припадки или изолированный эпилептический статус
|
||
|
4.1.3.
|
Приступы, связанные исключительно с острым воздействием метаболических или токсических факторов, а также депривация (лишение) сна, алкоголь, лекарства, эклампсия и т. д.
|
||
Классификация типов приступов ILAE 2017 г.
Рабочая группа по классификации типа приступов (Seizure Type Classification Task Force) была создана ILAE в 2015 г. Рабочая группа выбрала классификацию 1981 г. и ее последующие модификации в качестве основы для разработки обновленной Классификации. Основными мотивами для пересмотра классификации эпилептических приступов 1981 г. являлись следующие:
1. Ряд типов приступов, такие как тонические приступы или эпилептические спазмы, могут иметь либо очаговый, либо генерализованный дебют.
2. Отсутствие осведомленности о дебюте затрудняет классификацию приступов и представляет трудности для обсуждения в контексте классификации 1981 г.
3. Ретроспективное описание приступов часто не включает характеристику сохранения или изменения сознания пациента, которая, несмотря на то что является краеугольным камнем для многих типов приступов, представляет определенные сложности.
4. Ряд используемых терминов, таких как «психический», «парциальный», «простой парциальный», «сложный парциальный» и «дискогнитивный», не имеют ясного понимания у общества или высокого уровня признания у профессионального сообщества.
5. Ряд представляющих важность типов приступов не включены в классификацию поэтому типы приступов были классифицированы с учетом того, чтобы классификация была понятна всем, включая пациентов и членов их семей и применима к любому возрасту, включая новорожденных.
Используемая с 1981 г. структура классификации приступов была сохранена.
Следуя решениям 2010 г., использовался термин «фокальный» («очаговый») вместо термина «парциальный», поскольку данный термин более понятен в контексте определения локализации очага при начале приступа. Исключен термин «конвульсии» (“convulsion”), поскольку в ряде языков данный термин является синонимом термину «судороги» (“seizures”) а моторный контекст – недостаточно ясен. В обновленную классификацию добавлены термины «в сознании / сознание нарушено», «гиперкинетический», «когнитивный», «эмоциональный». Термин «в сознании / сознание нарушено» отражает способность осознавать себя и происходящее вокруг во время приступа. В Классификации приведены два термина, определяющих сознание – “awareness” и “consciousness”. Разработчики Классификации типов приступов ILAE 2017 г. указывают, что “impaired awareness” и “impairment of consciousness” являются синонимами, поэтому при адаптации классификации на русский язык “aware / impaired awareness” переведено как «в сознании / сознание нарушено». Термин «гиперкинетические приступы» добавлен в категорию фокальных приступов. Гиперкинетическая активность включает в себя ажитированные быстрые движения ногами, имитирующие удары или кручение педалей. Термин «когнитивный» заменил термин «психический» и относится к когнитивным расстройствам во время приступа, таким как афазия, апраксия или агнозия и к таким феноменам как дежавю, жамевю («никогда не виденное», противоположное дежавю состояние), иллюзии или галлюцинации. Термин «эмоциональный» обозначает эмоциональные проявления, такие как страх или радость. Также он применим к аффективным проявлениям эмоций при геластических или дакристических приступах (насильственный смех или плач).
Рабочая классификация приступов ILAE 2017 г. разработана в двух вариантах – в виде базовой и расширенной версии. На рисунке 1 представлена Базовая классификация приступов ILAE 2017 г., на рисунке 2 – Расширенная классификация приступов ILAE 2017 г. В базовой классификации не приведены подтипы судорожных приступов. Использовать можно как базовую, так и расширенную классификации, в зависимости от желаемой степени детализации. На первом этапе нужно определить, являются ли начальные проявления приступа фокальными или генерализованными. Если оценить начало приступа не представляется возможным, такой приступ относится к приступам с неуточненным дебютом.
Фокальные приступы с сохранением сознания соответствуют простым парциальным приступам. Фокальные приступы с нарушением сознания соответствуют сложным парциальным приступам. Нарушение сознания в течение любой из фаз фокального приступа дает основания отнести его к фокальным приступам с нарушением сознания.
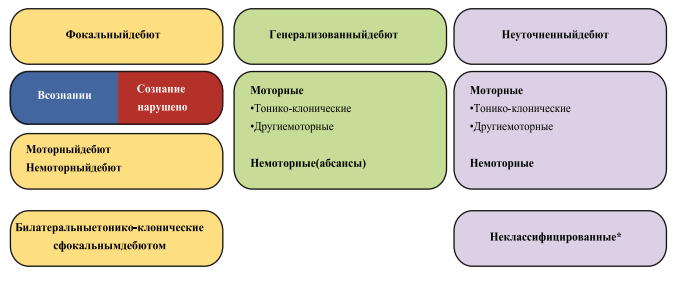
Рисунок 1.
Базовая классификация типов приступов Международной Противоэпилептической Лиги 2017 г.
Примечание. * Вследствие недостатка информации или невозможности отнести к другим категориям.
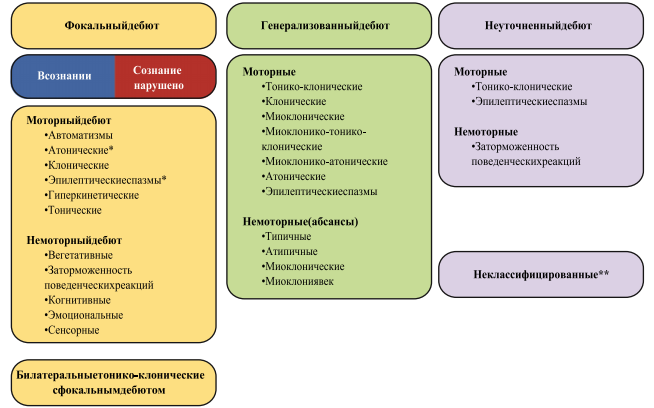
Рисунок 2.
Расширенная классификация типов приступов Международной Противоэпилептической Лиги 2017 г.
Примечания. * Состояние сознания обычно не определяется; ** вследствие недостатка информации или невозможности отнести к другим категориям.
Фокальные приступы с сохраненным сознанием или с нарушением сознания могут быть дополнительно охарактеризованы как приступы с моторным началом или немоторным началом, что отражает их первые проявления или симптомы.
При классификации фокального приступа допустимо исключить указания на состояние сознания в тех случаях, когда это неприменимо или состояние сознания неизвестно. В этом случае приступы классифицируют непосредственно на основании характеристик наличия/отсутствия движений в начале приступа.
Атонические судороги и эпилептические спазмы не имеют специфичных расстройств сознания.
Под когнитивными приступами подразумевают расстройство речи или других когнитивных функций – дежавю, галлюцинации, иллюзии и расстройства сознания.
Эмоциональные приступы включают тревогу, страх, радость, другие эмоции или наступление состояния аффекта без субъективных переживаний. Некоторые из компонентов эмоциональных приступов являются субъективными, поэтому должны быть уточнены совместно с пациентом или его опекуном.
Абсансы относятся к атипичным, если имеют медленное начало или завершение или же значительные изменения тонуса на фоне атипичных, медленных генерализованных спайк-волн на ЭЭГ.
Приступ является неклассифицируемым, если имеется недостаток информации или на основании доступной информации невозможно отнести приступ к тому или иному типу в других категориях.
Новыми типами фокальных приступов являются эпилептические спазмы, тонические, клонические, атонические и миоклонические приступы, которые ранее расценивались в качестве исключительно генерализованных приступов. Фокальные автоматизмы, вегетативные приступы, заторможенность поведенческих реакций, когнитивные, эмоциональные и гиперкинетические приступы являются новыми типами приступов, введенными в данную классификацию.
Вегетативные приступы сопровождаются ощущениями в желудочно-кишечном тракте, ощущением жара или холода, приливами, ощущением мурашек на коже, сердцебиением, сексуальным возбуждением, дыхательными расстройствами или другими вегетативными эффектами [32].
Билатеральные тонико-клонические приступы с фокальным началом также представляют собой новый тип приступов, их прежнее название – вторично-генерализованные приступы.
В обновленную классификацию введены новые типы генерализованных приступов, такие как абсансы с миоклонией век, миоклонико-атонические и миоклонико-тонико-клонические приступы. Миоклония век является наиболее значимым клиническим проявлением абсансов, они были помещены в категорию абсансов / немоторных приступов.
Эпилептические спазмы представляют собой судороги, которые можно отнести как к фокальным, так и к генерализованным, или неуточненным приступам, дифференциация может потребовать выполнение видео-ЭЭГ-мониторинга.
Фокальные приступы по своим двигательным проявлениям могут быть атоническими (фокальное снижение тонуса), тоническими (устойчивое фокальное увеличение тонуса), клоническими (фокальное ритмическое сокращение), или эпилептическими спазмами (фокальное сгибание или разгибание рук и сгибание туловища).
Различие между клоническими и миоклоническими приступами представляется несколько произвольным, но клонические приступы характеризуются устойчивыми, регулярными, отстоящими друг от друга во времени повторяющимися мышечными сокращениями, тогда как при миоклонусе сокращения менее регулярны и более краткие по времени.
Автоматизм представляет собой более или менее скоординированную повторяющуюся бесцельную двигательную активность. Для верификации фокального приступа с автоматизмами свидетелям приступа можно задать вопрос, демонстрировал ли пациент во время приступа повторяющиеся бесцельные фрагменты своего поведения, которые могли показаться нормальными в других обстоятельствах?
Фокальные моторные приступы с заторможенностью поведенческих реакций характеризуются прекращением двигательной активности и отсутствием ответной реакции. Поскольку краткое расстройство поведенческих реакций, встречающееся в начальной стадии многих типов приступов, является неспецифичным и трудно идентифицируемым.
Фокальные сенсорные приступы могут характеризоваться появлением обонятельных, зрительных, слуховых, вкусовых, вестибулярных ощущений, а также чувством жара или холода.
Классификация приступов с генерализованным дебютом схожа с классификацией 1981 г., но содержит несколько новых типов приступов. В качестве основного выбрано подразделение приступов на «моторные» и «немоторные (абсансы)». Когда название приступа однозначно указывает на наличие или отсутствие моторного компонента, например, «генерализованный тонический приступ», термин «моторный» или «немоторный» допустимо не применять. Также для таких приступов, которые могут быть только с генерализованным началом (например, абсансы), допустимо опускать термин «генерализованный».
Термин «тонико-клонические приступы» заменил ранее применявшийся французский термин “grand mal”. У тонико-клонических приступов начальной является именно тоническая фаза. Клоническая фаза тонико-клонического приступа обычно характеризуется мышечными подергиваниями с регулярно уменьшающейся частотой во время приступа. Во время тонико-клонического приступа нарушение сознания происходит до или одновременно с тонической или клонической фазами. В ряде случаев тонико-клонические приступы могут начинаться с неспецифических ощущений надвигающихся судорог или кратковременного изгиба головы или конечностей.
Начало, течение и завершение генерализованных клонических приступов характеризуется устойчивыми ритмическими подергиваниями головы, шеи, лица, туловища и конечностей с обеих сторон. Генерализованные клонические приступы встречаются гораздо реже, чем тонико-клонические приступы и обычно наблюдаются в младенчестве. Их следует отличать от беспокойства при тревожных состояниях и тремора при панических атаках.
Генерализованные тонические приступы проявляются в виде билатерального напряжения конечностей или их поднятия, часто вместе с напряжением мышц шеи. Тоническая активность может представлять собой устойчивую неестественную позу, возможно с растяжением или сгибанием частей тела, иногда сопровождающимся тремором конечностей. Тоническую активность трудно дифференцировать с дистонической активностью, которая характеризуется продолжительными сокращениями как мышц-агонистов, так и мышц-антагонистов, вызывающих атетоидные или «скручивающие» движения, которые могут приводить к неестественным позам.
Генерализованные миоклонические приступы могут возникать изолированно, либо в сочетании с тонической или атонической активностью. В отличие от клонуса при миоклонусе повторяющиеся движения более краткие и нерегулярные. Генерализованные миоклонико-тонико-клонические судороги начинаются с нескольких миоклонических подергиваний с последующей тонико-клонической активностью. Такие приступы наблюдаются у пациентов с ювенильной миоклонической эпилепсией, а иногда и с другими генерализованными эпилепсиями.
Миоклонико-атонический приступ характеризуется кратковременным подергиванием конечностей или туловища, за которым следует падение мышечного тонуса. Такие приступы, ранее называвшиеся миоклоническими-астатическими судорогами, чаще всего наблюдаются при синдроме Дуза, но также могут встречаться при синдроме Леннокса-Гасто и ряде других синдромов.
Атонические генерализованные приступы чаще сопровождаются падением пациента на ягодицы или вперед на колени и лицо. Восстановление обычно занимает несколько секунд. Типичным для тонических или тонико-клонических приступов, напротив, является падение назад.
Эпилептические спазмы ранее назывались инфантильными спазмами. Термин «инфантильные спазмы» остается релевантным для эпилептических спазмов, возникающих в младенческом возрасте. Эпилептический спазм представляет собой внезапное сгибание, растяжение или сочетание растяжения со сгибанием преимущественно проксимальных мышц или мышц туловища. Они обычно группируются в кластеры и встречаются чаще всего в младенческом возрасте.
В группу немоторных генерализованных приступов (абсансов) по-прежнему входят типичные и атипичные абсансы, эти два вида приступов ассоциированы с характерными изменениями на ЭЭГ, эпилептическими синдромами с соответствующей терапией и прогнозом. Согласно классификации 1981 г. абсансы следует отнести к атипичным, если наблюдаются нарушения тонуса, выраженные сильнее, чем при типичных абсансах, или начало и прекращение приступа не является внезапным. Для дифференциации между типичными и атипичными абсансами могут потребоваться данные ЭЭГ.
К миоклоническим абсансам относятся абсансы с ритмичными миоклоническими движениями с частотой 3 раза в сек., приводящие к удержанию в воздухе и постепенному поднятию верхних конечностей, ассоциированными с генерализованными пикволновыми разрядами с той же частотой на ЭЭГ. Миоклонические абсансы могут быть генетически-обусловленными, а также возникать без известных причин.
Миоклония век характеризуется миоклоническими подергиваниями век, девиацией глаз кверху, часто провоцируется светом или закрытием глаз. Миоклония век может ассоциироваться с абсансами, но также может представлять собой моторные приступы без абсансов, что затрудняет их классификацию. В классификации типов приступов ILAE 2017 г. миоклония век входит в группу немоторных приступов с генерализованным дебютом (абсансов). Абсансы с миоклонией век, судорогами и пароксизмальной активностью на ЭЭГ при закрывании глаз или световом воздействии составляют триаду синдрома Дживонса.
Для уточнения характера дебюта эпилептических спазмов необходимо проведение видео-ЭЭГ-мониторинга, это является важным для построения прогноза – приступы с фокальным дебютом могут лучше отвечать на терапию.
В Таблица №6 приведены термины, использующиеся в обновленной Классификации типов приступов ILAE 2017 г.
Таблица 6
|
Термин
|
Определение
|
Источник
|
|
Типичный абсанс
|
Внезапное начало, прерывание текущей активности, отсутствующий взгляд, возможна кратковременная девиация глаз. Обычно пациент не реагирует на обращение к нему. Продолжительность – от нескольких секунд до ½ мин. с очень быстрым восстановлением.
ЭЭГ демонстрирует генерализованные эпилептиформные разряды во время приступа (хотя метод не всегда доступен). Абсанс по определению является приступом с генерализованным дебютом. Термин не является синонимом «отсутствующего взгляда», который также может встречаться при судорожных приступах |
Адаптировано из [33]
|
|
Атипичный абсанс
|
Абсанс с изменениями в тонусе, которые являются более выраженными, чем при типичном абсансе; начало и/или прекращение, не являются внезапными, часто ассоциирован с медленной нерегулярной генерализованной пик-волновой активностью на ЭЭГ
|
Адаптировано из [34]
|
|
Атонический (приступ)
|
Внезапная потеря или снижение мышечного тонуса без видимого предшествующего миоклонического или тонического компонента длительностью ~ 1-2 сек., включая мышцы головы, туловища, лица или конечностей
|
[33]
|
|
Автоматизм
|
Более или менее скоординированная двигательная активность, которая обычно возникает на фоне расстройства когнитивных функций, чаще с последующей амнезией. Часто напоминает контролируемые движения и может представлять собой измененную двигательную активность, имевшую место до приступа
|
[33]
|
|
Вегетативные (автономные) приступы
|
Явное изменение функции вегетативной нервной системы, включающее изменение диаметра зрачков, потоотделение, изменение тонуса сосудов, терморегуляции, расстройства функции ЖКТ и сердечно-сосудистой системы
|
Адаптировано из [33]
|
|
Аура
|
Внезапный субъективный феномен, специфичный для конкретного пациента, который может предшествовать приступу
|
[33]
|
|
Сознание
(«Aware») |
Осознавание себя или способность ориентироваться в окружающем пространстве
|
Новый
|
|
Билатеральный
|
Вовлекающий левую и правую стороны, хотя проявления билатеральных приступов могут быть как симметричными, так и асимметричными
|
Новый
|
|
Клонический (приступ)
|
Подергивание, симметричное или асимметричное, которое регулярно повторяется и включает одни и те же группы мышц
|
Адаптировано из [33]
|
|
Когнитивный
|
Относится к мышлению и высшим корковым функциям, таким как язык, пространственное восприятие, память и праксис. Предыдущий термин для аналогичного использования в контексте определения типа приступов был «психический»
|
Новый
|
|
Сознание («Consciousne ss»)
|
Как субъективные, так и объективные аспекты состояния рассудка, включающие осознание себя как уникальной сущности, восприятие, ответные реакции и память
|
Новый
|
|
Дакристический (приступ)
|
Сопровождающийся эпизодами плача, который не обязательно может быть связан с грустью
|
[33]
|
|
Дистонический (приступ)
|
Сопровождающийся устойчивыми сокращениями как агонистических, так и антагонистических мышц, вызывающих атетоидные или скручивающие движения, которые могут вызывать неестественные позы
|
Адаптировано из [33]
|
|
Эмоциональные приступы
|
Приступы с эмоциями или появлением эмоции как ранней характерной черты, такие как страх, спонтанная радость или эйфория, смех (геластический) или плач (дакристический)
|
Новый
|
|
Эпилептические спазмы
|
Внезапное сгибание, растяжение или чередование сгибания и растяжения преимущественно проксимальных мышц и мышц туловища, которое обычно более длительное, чем миоклоническое, но не такое длительное, как тонический приступ. Могут возникнуть гримасы, кивки головы или мелкие движения глаз. Эпилептические спазмы часто развиваются в виде кластеров. Инфантильные спазмы в младенчестве являются наиболее известной формой, но эпилептические спазмы могут возникать в любом возрасте
|
Адаптировано из [33]
|
Термины, использующиеся в обновленной классификации типов приступов ILAE 2017 г.
|
Эпилепсия
|
Болезнь мозга, определяемая любым из следующих условий: (1) по крайней мере, два неспровоцированных (или рефлекторных) приступа, с интервалом >24 ч; (2) один неспровоцированный (или рефлекторный) приступ и вероятность повторения приступов, близкая к общему риску рецидива (≥60%) после двух спонтанных приступов, в по- следующие 10 лет; (3) диагноз эпилептического синдрома.
Эпилепсия считается разрешившейся у достигших определенного возраста пациентов с зависящим от возраста эпилептическим синдромом либо при отсутствии эпилептических приступов в течение 10 лет. У пациентов, не использовавших антиэпилептические препараты (АЭП) не менее 5 лет |
[13]
|
|
Миоклония век
|
Подергивание век с частотой не менее 3 раз в сек., как правило, с девиацией глаз вверх, длящееся как правило <10 сек., часто провоцируется закрытием глаз. В части случаев может сопровождаться кратковременной потерей ориентации
|
Новый
|
|
Припадок фехтовальщика
|
Тип фокального моторного приступа с вытягиванием одной руки и сгибанием в локте другой, имитирующий фехтование с рапирой. Также называется «судороги дополнительной моторной зоны»
|
Новый
|
|
Приступы
«цифра 4» |
Приступы, характеризующиеся разгибанием одной руки перпендикулярно туловищу (обычно контралатеральной эпилептогенной зоне в головном мозге) и сгибанием в
локте другой руки, образующими цифру «4» |
Новый
|
|
Фокальный (приступ)
|
Возникающий в сетевых структурах, ограниченных одним полушарием. Он может быть дискретно локализован или иметь более широкое распространение. Фокальные приступы могут возникать в подкорковых структурах
|
[35]
|
|
Билатеральные тонико-клонические приступы с фокальным
дебютом |
Тип приступов с фокальным дебютом, с сохранением или нарушением сознания, могут быть моторными или немоторными, затем характеризующиеся развивающейся билатеральной тонико-клонической активностью. Предыдущий термин – «вторично-генерализованные приступы с парциальным дебютом»
|
Новый
|
|
Геластический (приступ)
|
Взрывы смеха или хихиканья, обычно без соответствующего аффективного фона
|
[33]
|
|
Генерализованный (приступ)
|
Первоначально возникающий одномоментно, с быстрым вовлечением билатерально расположенных сетевых структур
|
[35]
|
|
Миоклонический (приступ)
|
Внезапное, краткое (<100 мс) непроизвольное одиночное или множественное сокращение мышц или групп мышц с переменной топографией (аксиальная, проксимальная, мышцы туловища, дистальная). При миоклонусе движения повторяются менее регулярно и с меньшей продолжительностью, чем при клонусе
|
Адаптировано из [33]
|
|
Миоклонико- атонический
|
Генерализованный тип приступов с миоклоническими подергиваниями, предшествующими атоническому моторному компоненту. Этот тип приступов ранее назывался миоклонико-астатическим
|
Новый
|
|
Миоклоникотонико-клонический
|
Одно или несколько билатеральных подергиваний мышц туловища, с последующим развитием тонико-клонического приступа. Первоначальные подергивания можно рассматривать как короткий период клонуса или миоклонуса. Приступы такого типа характерны для ювенильной миоклонической эпилепсии
|
Выделено из
[34] |
|
Немоторный
|
Фокальный или генерализованный приступ, при которых не проявляется двигательная (моторная) активность
|
Новый
|
|
Распространение
|
Распространение судорожной активности из одного мозгового центра на другой или вовлечение дополнительных сетевых структур мозга
|
Новый
|
|
Ответная реакция
|
Способность адекватно отреагировать движением или речью на предъявленный стимул
|
Новый
|
|
Приступ
|
Преходящее появление признаков и/или симптомов, связанных с аномальной избыточной или синхронной активностью нейронов в головном мозге
|
[14]
|
|
Сенсорный
приступ |
Субъективно воспринимаемое ощущение, не вызванное соответствующими стимулами во внешнем мире
|
[33]
|
|
Спазм
|
См. «эпилептический спазм»
|
|
|
Тонический (приступ)
|
Устойчивое нарастающее сокращение мышц продолжительностью от нескольких секунд до нескольких минут
|
[33]
|
|
Тонико-клонический (приступ)
|
Последовательность, состоящая из фазы тонического сокращения, за которой следует клоническая фаза
|
[33]
|
|
Бессознательный
|
Термин «бессознательный» может использоваться в качестве сокращенного термина «нарушение сознания»
|
Новый
|
|
Неклассифицированный
|
Может применяться по отношению к типу приступов, который не описан в Классификации ILAE 2017 г. по причине недостаточной информации либо необычных клинических признаков. Если приступ не классифицирован в связи с недостаточной информацией о его начале, он может быть ограниченно классифицирован на основании доступных для интерпретации данных.
|
Новый
|
|
Отсутствие ответа
|
Неспособность адекватно отреагировать движением или речью на предъявляемый стимул
|
Новый
|
|
Версивный (приступ)
|
Длительное насильственное сопряженное вращение глаз, головы и туловища или их отклонение латерально от центральной оси
|
[33]
|
Таблица 7(продолжение)
Генно-картированные эпилептические синдромы
|
Эпилептический синдром
|
Ген
|
Локус
|
|
Эпилепсия с генерализованными тонико-клоническими судорогами при пробуждении
|
CLCN2, EGMA, ECA3 |
3q26-qter |
|
Доброкачественные семейные неонатальные судороги, 2-го типа
|
KCNQ3, EBN2,
BFNC2 |
8q24
|
|
Доброкачественные семейные неонатальные судороги, 1-го типа
|
KCNQ2, EBN1
|
20q13.3
|
|
Детская абсансная эпилепсия
|
ECA1
|
8q24
|
|
Детская абсансная эпилепсия
|
GABRG2, GEFSP3, CAE2, ECA2
|
5q31.1‒q33.1
|
|
Детская абсансная эпилепсия
|
CLCN2, EGMA, ECA3
|
3q26‒qter
|
|
Эпилепсия у женщин с умственной отсталостью
|
EFMR
|
Xq22
|
|
Генерализованная идиопатическая эпилепсия
|
CACNB4
|
2q22‒q23
|
|
Генерализованная идиопатическая эпилепсия
|
EGI
|
8q24
|
|
Генерализованная эпилепсия с фебрильными приступами плюс
|
GABRG2, GEFSP3, CAE2, ECA2
|
5q31.1‒q33.1
|
|
Генерализованная эпилепсия с фебрильными приступами плюс 2-го типа
|
SCN1A, GEFSP2 |
2q24 |
|
Ювенильная абсансная эпилепсия
|
CLCN2, EGMA, ECA3
|
3q26‒qter
|
|
Ювенильная абсансная эпилепсия
|
EJM1
|
6p
|
|
Ювенильная абсансная эпилепсия
|
EJM2, JME
|
15q14
|
|
Ювенильная абсансная эпилепсия
|
CACNB4
|
2q22‒q23
|
|
Ювенильная абсансная эпилепсия
|
CLCN2, EGMA, ECA3
|
3q26‒qter
|
|
Ювенильная абсансная эпилепсия 2-го типа
|
GABRA1
|
5q34‒q35
|
|
Ювенильная миоклоническая эпилепсия Лафора
|
EPM2A, MELF, EPM2
|
6q24
|
|
Ювенильная миоклоническая эпилепсия Лафора
|
NHLRC, EPM2B
|
6p22.3
|
|
Доброкачественная семейная миоклоническая эпилепсия у взрослых
|
MEBA, BAFME, FAME
|
8q24
|
|
Ночная лобная эпилепсия
|
CHRNB2, EFNL3
|
1q21
|
|
Ночная лобная эпилепсия 2-го типа
|
ENFL2
|
15q24
|
|
Парциальная эпилепсия со слуховыми симптомами
(аутосомно-доминантная височная эпилепсия) |
LGI1, EPT |
10q24 |
|
Эпилепсия с парциальными перицентральными спайками
|
EPPS
|
4p15
|
|
Семейная парциальная эпилепсия с вариабельным фокусом
|
FPEVF
|
22q11‒q12
|
|
Прогрессирующая миоклонусэпилепсия
|
CSTB, STFB, EPM1
|
21q22.3
|
|
Пиридоксин-зависимая эпилепсия
|
EPD, PDE
|
5q31.2‒q31.3
|
Для облегчения использования классификации типов приступов ILAE 2017 г. практикующими специалистами был разработан следующий алгоритм, содержащий советы экспертов по очередности действий и критериям оценки приступов.
-
Начало: определите, относится ли приступ к фокальным или генерализованным с использованием 80%-го уровня достоверности. При уровне достоверности <80% начало следует расценивать как неуточненное.
-
Сознание: при фокальных судорогах необходимо решить, следует ли классифицировать по состоянию сознания или же отказаться от использования критерия сознания при классификации. «Фокальные приступы с сохранением сознания» соответствуют «простым парциальным приступам», «фокальные приступы с нарушением сознания» – «сложным парциальным приступам» в старой терминологии.
-
Нарушение сознания в любой момент: фокальный приступ является «фокальным приступом с нарушением сознания», если сознание нарушается в любой момент приступа.
-
Принцип доминирования дебюта: классифицировать фокальный приступ необходимо, принимая во внимание первый признак или симптом (за исключением заторможенности поведенческих реакций).
-
Заторможенность поведенческих реакций: при «фокальном приступе с заторможенностью поведенческих реакций» заторможенность поведенческих реакций является характерной чертой всего приступа.
-
Моторный/немоторный: «фокальный приступ с сохраненным сознанием» или «фокальный приступ с нарушенным сознанием» может быть дополнительно классифицирован на основании характеристик двигательной (моторной) активности. И наоборот, фокальный приступ может классифицироваться на основании характеристик двигательной (моторной) активности без указания состояния сознания, например, «фокальный тонический приступ».
-
Необязательные термины: ряд определений, такие как моторный или немоторный могут быть опущены, если тип приступов однозначно указывает на них.
-
Дополнительные характеристики: после классификации типа приступов, основанной на начальных проявлениях, рекомендуется добавлять описания других признаков и симптомов из числа предлагаемых характеристик или в свободной форме. Дополнительные характеристики не могут изменять тип приступов. Пример: фокальный эмоциональный приступ с тонической активностью правой руки и гипервентиляцией.
-
Билатеральный или генерализованный: необходимо использовать термин «билатеральный» для тонико-клонических приступов, которые распространяются на оба полушария и «генерализованный» при приступах, которые, по-видимому, изначально развиваются одновременно в обоих полушариях.
-
Атипичный абсанс: абсанс является атипичным, если он имеет медленное начало или завершение, заметные изменения тонуса или спайк-волны <3 Гц на ЭЭГ.
-
Клонический или миоклонический: продолжительные ритмические подергивания относятся к клоническим и регулярные непродолжительные подергивания относятся к миоклонии.
-
Миоклония век: абсанс с миоклонией век представляет собой насильственные подергивания век во время абсансного приступа.
Классификация эпилепсии ILAE 2017г
Классификация эпилепсии ILAE 2017 г. является многоуровневой и предназначена для применения в клинической практике (см. рис. 3). Различные уровни классификации – тип приступов, тип эпилепсии и эпилептический синдром – предусмотрены в связи с тем, что у практикующих специалистов, широко разниться доступ к необходимым ресурсам, таким как средства инструментальной диагностики. Там, где это возможно, ILAE рекомендует устанавливать диагноз на всех трех уровнях и дополнительно устанавливать этиологию эпилепсии.
Второй уровень Классификации эпилепсии ILAE 2017 г. подразумевает опре- деление типа эпилепсии. В дополнение к уже широко применяющимся определениям «фокальная эпилепсия» и «генерализованная эпилепсия» в Классификацию эпилепсии ILAE 2017 г. введен новый тип «комбинированная генерализованная и фокальная эпилепсия», а также категория «Неуточненная эпилепсия». Фокальная эпилепсия включает в себя однофокальные и мультифокальные расстройства, а также приступы с вовлечением одного полушария. При фокальной эпилепсии могут наблюдаться та- кие типы приступов, как фокальные с сохранением сознания, фокальные с нарушением сознания, фокальные моторные приступы, фокальные немоторные приступы, билатеральные тонико-клонические приступы с фокальным дебютом. На интериктальной ЭЭГ обычно регистрируются фокальные эпилептиформные разряды. Вместе с тем, диагноз следует ставить на основании клинических данных, используя результаты ЭЭГ в качестве дополнительных данных.
Диагноз генерализованной эпилепсии ставится на основании клинических данных, подтвержденных результатами ЭЭГ, где имеются типичные разряды, регистрирующиеся между приступами – генерализованная пик-волновая активность.
В Классификацию эпилепсии ILAE 2017 г. введена новая группа комбинированных генерализованных и фокальных эпилепсий, поскольку существуют пациенты, которые имеют как генерализованные, так и фокальные эпилептические приступы. Диагноз также ставится на основании клинических данных, подтвержденных результатами ЭЭГ. Интериктальная ЭЭГ может содержать генерализованную пик-волновую активность, но наличие эпилептиформной активности не является необходимым для постановки диагноза.
Тип эпилепсии также может быть конечным уровнем детализации диагноза, что допустимо в тех случаях, когда клиницист не может определить эпилептический синдром.
Термин «неуточненная эпилепсия» используется в тех случаях, когда есть понимание наличия у пациента эпилепсии, но врач не может определить, является ли тип эпилепсии фокальным или генерализованным, потому что не располагает достаточной информацией. Возможно, нет доступа к ЭЭГ, или ЭЭГ-исследование оказалось неинформативным. Если тип приступов является неуточненным, то тип эпилепсии также может быть неуточненным по тем же причинам, хотя эти два определения не обязательно должны быть согласованы между собой.
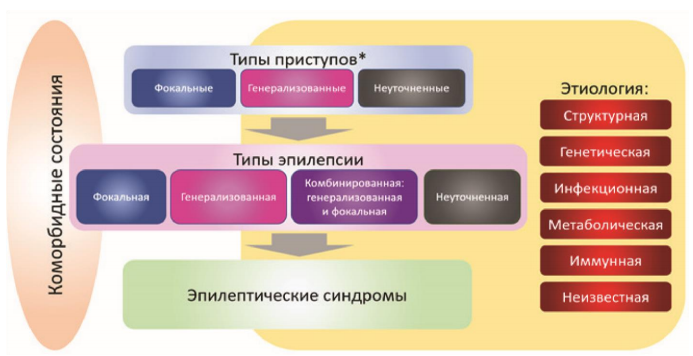
Рисунок 3.
Структура Классификации эпилепсии ILAE 2017 г.
Примечание. * Оценивается по началу приступа. Третий уровень – это диагностика синдрома эпилепсии. Синдром представляет собой группу признаков, включающих типы приступов, ЭЭГ и находки при нейровизуализации, которые имеют тенденцию сопутствовать друг другу. Определение синдрома важно для уточнения этиологии, лечения и прогноза. Многие синдромы, такие как детская абсансная эпилепсия, синдром Уэста и синдром Драве хорошо известны.
В перечне приводятся наиболее часто встречаемые эпилепсии и эпилептические синдромы у детей.
1.1. Ранняя младенческая энцефалопатия с супрессивно-взрывным паттерном на ЭЭГ (синдром Отахара).
1.2. Тяжелая эпилепсия с множественными независимыми фокусами спайков (SEMISE, синдром Марканда — Блюме — Отахара).
1.3. Тяжелая миоклоническая эпилепсия младенцев (Драве).
1.4. Злокачественные мигрирующие парциальные приступы младенчества (MMPSI- синдром Коппола — Дулока).
1.5. Синдром Веста.
2. Эпилептические энцефалопатии детского возраста:
2.2. Синдром Леннокса — Гасто.
2.3. Синдром гемиконвульсивных приступов, гемиплегии, провоцируемый гипертермией, эпилепсии (синдром IННЕ).
2.4. Разрушительная эпилептическая энцефалопатия у детей школьного возраста, вы- званная гипертермией (FIRES-синдром, ранее называемый DESC-синдром).
3. Эпилептические энцефалопатии, сопровождающиеся постоянной продолжительной эпилептической активностью в фазе медленного сна на ЭЭГ:
3.2. Когнитивная эпилептиформная дезинтеграция с эпилептической активностью на ЭЭГ и без эпилептических приступов.
4. Эпилептические энцефалопатии при наследственных заболеваниях и хромосомных аномалиях:
4.2. Синдром Миллера — Дикера — лиссэнцефалия: краниофациальный дисморфизм, микроцефалия, широкая переносица, укороченные пальцы.
4.4. Синдром Ангельмана — лицевой дисморфизм, широкие зубы, гипопигментация кожи, гипотония, «счастливая марионетка».
4.5. Туберозный склероз (инфантильные спазмы, синдром Веста, эпилепсия).
4.6. Синдром Прадера — Вилли — мышечная гипотония, гипогенитализм, крипторхизм, синдактилия, выворот век.
4.7. MERRF — миоклоническая (митохондриальная) эпилепсия красных разорванных волокон.
Таблица 10
| Вид ЭЭ | Известные гены, мутации в которых приводят к их развитию | Пол | Возраст дебюта | Вид эпилептических приcтупов | ЭЭГ | Лечение | Эволюция эпилепсии, исход | Дифференциальный диагноз | Когнитивные нарушения |
| Ранняя младенческая ЭЭ (синдром Отахара) | STXBP1 (30%), KCNQ2 (20%), SCN2A (10%), AARS, ARX, BRAT1, CACNA2D2, GNAO1, KCNT1, NECAP1, PIGA, PIGQ, SCN8A, SIK1, SLC25A22 | М=Ж, кроме ARX – болеют в основном мальчики | 0 –3 месяца | Тонические приступы, могут быть фокальные и инфантильные спазмы, редко – миоклонус | Интериктальная активность: супрессивно-взрывные изменения. Иктальная: диффузное замедление или быстрая низковольтажная активность, фокальные спайки, гипсаритмия | Фармакорезистентность, может быть эффективна кетогенная диета | В 75% случаев трансформация в синдром Веста. Приступы продолжаются на протяжении жизни | Ранняя миоклоническая ЭЭ | Выраженная степень |
| Ранняя миоклоническая ЭЭ | ERBB4, PIGA, SETBP1, SIK1, SLC25A22 | М=Ж | 0 –3 месяца |
Фрагментарный
миоклонус, реже – тонические и фокальные приступы |
Интериктальная активность: супрессивно-взрывные изменения, ухудшающиеся во время сна. Иктальная: паттерн миоклонуса | Фармакорезистентность |
В большинстве случаев не эволюционирует в другие виды ЭЭ, сохраняются
миоклонус и фокальные приступы
|
Ранняя младенческая ЭЭ | Выраженная степень |
| ЭЭ младенчества с мигрирующими фокальными приступами | KCNT1 (50%), SCN2A (25%), PLCB1, QARS, SCN1A, SCN8A, SLC25A22, TBC1D24, SLC12A5 | М=Ж | 0 – 6 месяца | Фокальныеные приприступы ступы, исходящие из различных независимых очагов в обеих гемисферах | Интериктальная: может быть нормальной в дебюте, далее мульти-фокальная активность. Иктальная: мигрирующие фокусы | В большинстве случаев – фармакорезистентность, может быть эффективны фенитоин, кетогенная диета | 7% инфанальные спазмы часто сохраняются фокальные приступы после 1 года | Ранняя младенческая ЭЭ; ранняя миоклоническая ЭЭ | До начала приступов может быть нормальное развитие, после дебюта – регресс развития. |
| Синдром Веста | CDKL5(10%), STXBP1 (2%), ARX, ALG13, DOCK7, DNM1, FOXG1 | М=Ж, при ARX страдают мальчики, при CDKL5 – чаще девочки | 2 –12 месяцев | Эпилептические спазмы | Интериктальная: гипсаритмия. Иктальная: электро-декремент | Терапия первой линии: кортикостероиды, вигабатрин, кетогенная диета | Может быть трансформация в синдром Леннокса – Гасто | До начала приступов может быть нормальное развитие, после дебюта – регресс развития. |
| Вид ЭЭ | Известные гены, мутации в которых приводят к их развитию | Пол | Возраст дебюта | Вид эпилептических приступов | ЭЭГ | Лечение | Эволюция эпилепсии, исход | Дифференциальный диагноз | Когнитивные нарушения |
| Синдром Драве | SCN1A (90%), PCDH19, GABRA1, GABRG2, HCN1, STXBP1 | М=Ж, при PCDH19 – де-вочки (99%) | 5–16 месяцев | Фебрильные и афебрильные гемиклонические и генерализованные тонико-клонические приступы, часто эпилептический статус | Нормальная ЭЭГ в течение 1-2лет, позже генерализованные или мульти-фокальная эпилептиформная активность, часто фотосенситивность | Фармакорезистентность наилучший эффект – топиромат, стирипентол в комбинации с вальпроевой кисло-той, клобазам, левитирацетам, кетогенная диета. Противопоказаны: карбамазепин, ламотриджин | Сохраняющиеся приступы: от 1 до 5 лет – фокальные, миоклонические, абсансы, неконвульсивный эпилептический статус. После 5 лет – короткие ночные приступы, легкий миоклонус | Эпилепсия с миоклонически-атоническими приступами, синдром Леннокса – Гасто | Нормальное развитие в 1 год жизни, замедленное – между 1 и 2 годами, регресс после эпилептических статусов. Исход: от легких до тяжелых когнитивных нарушений |
| Эпилепсия с миоклонически-атоническими приступами | SLC2A1 (5%), SLC6A1 (4%), CHD2, GABRA1, GABRG2, SCN1A, SCN1B, KCNA2 | М:Ж=2:1 | 7 месяцев – 6 лет | Миоклонически-атонические приступы, абсансы, тонико-клонические приступы, неконвульсивный эпилептический статус | Иктальная: замедление тета- и дельта-диапазона, генерализованные спайк-полиспайк-волны. В некоторых случаях – фотосенситивность | Фармакорезистентность часто, может быть эффективна кетогенная диета (50%), кортикостероиды | У большинства пациентов – ремиссия через 3–5 лет от начала, в тяжелых случаях – сохранение приступов (преимущественно ночных тонических) | Доброкачественная миоклоническая эпилепсия младенчества, синдром Леннокса – Гасто | Раннее развитие – норма. Регресс часто наблюдается после дебюта эпилепсии. Ис-ход от нормального интеллекта (26–67%) до тяжелого расстройства |
| Синдром Леннокса–Гасто | ALG13, CACNA1A, CDKL5, CHD2, DNM1, FLNA, GABRB3, GRIN2B, HNPRNU, HNRNPH1, IQSEC1, iQSEC2, KCNQ3, MTOR, SCN1A, SCN2A | М=Ж | 1–8 лет | Тонические приступы с/без атипичными абсансами, атоническими, миоклоническими или генерализованными тонико-клоническими приступами. Эпизоды тонического или неконвульсивного эпилептического статуса | Интериктальная: замедление основной активности, спайк-волны <2–5 Гц, генерализова-ная пароксизмальная быстрая активность во сне. Иктальная: электродекремент, низковольтажная быстрая активность, медленные спайк-волны, генерализованные спайк-полиспайк волны | Фармакорезистентность | У 80% при-ступы сохраняются во взрослом возрасте | Эпилепсия с миоклонически-атоническими приступами, синдром Драве | Нарушение интеллекта до начала приступов наблюдается у 20–60%, к 5 годам 90% имеют когнитивные нарушения |
| Эпилептическая афазия: синдром Ландау–Клеффнера | GRIN2А (10-20%) | Не известно | 3–7 лет | Роландические эпилептические приступы, могут быть негативный миоклонус и атонические приступы | Электрический статус медленного сна (>85% non-REM сна) |
Может быть фармакорезистентность.
Препараты выбора: кортикостероиды, бензодиазепин
|
Эпилепсия является возрастзависимым состоянием, приступы заканчиваются к подростковому возрасту | Синдром Леннокса – Гасто | Развитие до приступов в норме. Исход: нарушение речи от легкого до тяжелого |
Таблица 11
| Патология | Возраст начала | Этиология | Тип судорог | Характеритика ЭЭГ | Потеря функции |
| Ранняя инфантильная эпилептическая энцефалопатия/Ранняя миоклоническая энцефалопатия | Неонатальный период (до 3 месяцев) | Пороки развития мозга, нарушения обмена веществ, генетические мутации | Тонические судороги (EIEE), миоклонические (EME) и другие виды | Схема подавления взрыва | Серьезные нарушения развития, переход к другим типам эпилептических энцефалопатий (инфантильные спазмы, синдром Леннокса-Гасто) |
| Инфантильные спазмы | Младенчество (3-7 месяцев) | Неизвестно (30%), известно (70%; генетические, структурные, метаболические нарушения) | Эпилептические спазмы | Гипсаритмия (интериктальная) и электродекрементальная реакция (иктальная) | Задержка развития, эволюция к синдрому Леннокса-Гасто |
| Синдром Драве | В течение 1 года жизни | Генетические мутации (SCN1A) | Фебрильные судороги на начальном этапе, генерализованные тонико-клонические, очаговые и другие типы позже | Изначально нормально. Эволюция к генерализованным спайко-волновым и фокальным эпилептиформным разрядам позже | Задержка развития |
| Синдром Ленокса-Гасто | Период детства (1-7 лет) | См “инфантильные спазмы” | Тонические, атонические, миоклонические, атипичные абсансы и др. | Генерализованный медленный спайк-волновой (interictal) | Задержка развития |
| СЭС | Период детства (пик в 4-8 лет) | Неизвестные генетические мутации (GRIN2A) | Фокальные клонические, абсансы, генерализованные тонико-клонические | Непрерывный всплеск во время медленного сна | Задержка когнитивных функций и нарушение поведения; домен может отражать расположение эпилептиформным разрядам |
| Синдром Ландау Клефнера | Период детства (примерно в 5-7 лет) | Неизвестные генетические мутации (GRIN2A) | Похож на СЭС, но может не иметь судорог | Некоторые имеют непрерывный всплеск во время медленного сна | Приобретенная афазия (слуховая агнозия) |
| Аутоиммунная эпилепсия | От младенчества до периода взрослой жизни | Аутоантитела (анти-NMDAR-антитела, анти нейрональные белковые анти-тела) могут быть обнаружены | Генерализованные тонико-клонические, очаговые, эпилептический статус | Генерализованные или очаговые эпилептиформные разряды | Задержка когнитивных функций и нарушение поведения (может быть обратимым при иммунотерапии) |
Этиология и патогенез
Этиология и патогенез
Таблица 8
Основные этиологические факторы эпилепсии
|
Генетические отклонения
|
Этиологические факторы эпилепсии
|
|
Дисгенетические аномалии
|
Ассоциированные как с генетическими нарушениями, так и с ранним онтогенетическим повреждением головного мозга
|
|
Инфекции
|
Менингит (вирусный, бактериальный, грибковый и паразитарный). Энцефалит (наиболее часто вирус простого герпеса, различные арбовирусы и определенные хронические энцефалиты). Абсцессы (бактериальной, грибковой или паразитарной этиологии)
|
|
Черепно-мозговые травмы
|
Ранения твердой мозговой оболочки, внутричерепные гематомы, генетическая предрасположенность. Для развития эпилепсии необходим «процесс созревания». Дебют эпилепсии посттравматического генеза наиболее часто наблюдается в срок до двух лет. Риск эпилепсии в 12 раз выше, чем в общей популяции, где он составляет 0,5‒1,0 %. Факторами, увеличивающими риск возникновения приступов, являются структурные повреждения, глиозные рубцы, геморрагические осложнения
|
|
Цереброваскулярные заболевания
|
Постинсультные приступы: ранние возникают в 3‒9 % случаев, поздние ‒ в 3‒5 % случаев и чаще наблюдаются после ишемических инсультов
|
|
Опухоли
|
При медленно растущих доброкачественных опухолях чаще возникают эпилептические приступы. Кортикальные дисгенезии и гиппокампальные аномалии во многом сходны с дисгенетическими опухолями, такими как дисэмбриопластические: нейроэпителиома и ганглиома
|
|
Гиппокампальный склероз
|
Представлен в основном атрофией и глиозом (характеризуется повышением интенсивности сигнала в режимах FLAIR и Т2) и может быть выявлен при мезиальной височной эпилепсии
|
|
Каверномы
|
Сосудистые мальформации с генетической предрасположенностью к развитию эпилепсии. Наиболее часто встречается в виде многоочагового поражения
|
|
Метаболические нарушения и токсическая зависимость
|
Главным образом, они вызывают острые симптоматические приступы. Наиболее важными являются заболевания с нарушением метаболизма глюкозы и кальция. Также определенную роль играют гормональные нарушения
|
Таблица 9
Наследственные болезни обмена, сочетающиеся с эпилепсией, в зависимости от возраста дебюта заболевания
|
Возраст дебюта
|
Наследственные болезни обмена с эпилепсией
|
|
Неонатальный
|
Гипогликемия, пиридоксин-зависимые приступы, некетогенная гиперглицинемия, органические ацидурии, нарушение цикла мочевой кислоты (гипераммонемия), неонатальная адренолейкодистрофия, синдром Целльвегера, приступы, зависимые от фолиевой кислоты, дефицит синтеза голокарбоксилазы, дефицит кофактора молибдена, дефицит сульфат оксидазы
|
|
Младенческий
|
Гипогликемия, нарушение транспорта глюкозы (GLUT1 недостаточность), недостаточность креатина, биотинидазная недостаточность, аминоацидопатии, органические ацидурии, наследственные нарушения гликозилирования, пиридоксинзависимые приступы, инфантильная форма нейронального цероидного липофусциноза (NCL 1)
|
|
Ранний детский
|
Поздняя инфантильная форма нейронального цероидного липофусциноза (NCL 2), митохондриальные заболевания (включая болезнь Альперса), лизосомальные болезни накопления
|
|
Школьный
|
Митохондриальные заболевания, ювенильная форма нейронального цероидного липофусциноза (NCL 3), прогрессирующие формы эпилепсии с миоклонусом
|
Основную роль в патогенезе эпилепсии играет нарушение функции нейронов, которая связана с патологией нейрональных мембран и дисбалансом между возбуждающими и тормозящими системами головного мозга.
Каждый нейрон имеет потенциал покоя от –50 до –80 мВ (милливольт), выражающийся в разнице вольтажа между наружной и внутренней поверхностью клетки. Разность потенциалов (поляризация) существует благодаря тому, что положительно и отрицательно заряженные частицы разделены клеточной мембраной. Мембрана пронизана каналами, осуществляющими транспорт ионов внутрь и вовне. Основные ионы, определяющие поляризацию, – Na, Ca, Cl, K. Изменения проницаемости мембраны, позволяющие ионам натрия и кальция проникать в клетку, приводят к деполяризации. Изменения мембраны, позволяющие калию покидать клетку или ионам хлора проникать в нее, приводят к гиперполяризации. Уровень поляризации мембраны в –20 мВ является критическим, и после превышения его нейрон генерирует потенциал действия – спайк. Эпилептический нейрон характеризуется нестабильностью мембраны и выраженной тенденцией к ее деполяризации. Возникающая спонтанно или под влиянием афферентных стимулов деполяризация мембраны с соответствующим нарастанием готовности к генерации потенциалов действия называется пароксизмальным деполяризационным сдвигом, что является основой эпилептогенеза на уровне отдельно взятого нейрона.
Происходящие процессы могут быть связаны не только с нарушениями нейрохимических процессов на мембранном уровне, но и с патологией синаптических взаимодействий через специфические возбудительные и тормозные рецепторы. В ответ на воздействие нейромедиаторов немедленно изменяется проницаемость ионных каналов. Активация с помощью возбуждающих нейромедиаторов (глутамат, аспартат) соответствующих синапсов приводит к возникновению возбуждающего постсинаптического потенциала и деполяризации мембраны. Активация тормозного синапса с помощью гамма-аминомасляной кислоты (основного тормозного нейромедиатора) приводит к возникновению тормозного постсинаптического потенциала и гиперполяризации мембраны. Принимающая нервная клетка исправно фиксирует эти противоположные импульсы, которые определяют ее собственное действие. Если возбуждающие воздействия поступают на нейрон с короткими интервалами или одномоментно через многие синапсы, то они суммируются, деполяризация оказывается пропорционально выше и продолжительнее, что определяет высокую частоту и продолжительность генерации этими нейронами потенциалов действия, которые распространяются к другим нейронам и усиливают в них возбуждение. Помимо нейротрансмиттеров важную роль в эпилептогенезе и его торможении играют нейропептиды, нейромодуляторы. Они изменяют чувствительность синаптических рецепторов мембраны к нейротрансмиттерам, регулируют готовность пресинаптической бляшки выбрасывать нейротрансмиттер, активность обратного захвата или разрушения нейромедиатора.
Развитие эпилепсии может быть обусловлено двумя принципиально различными патофизиологическими механизмами, которым соответствует очаговая и генерализованная пароксизмальная эпилептическая активность, проявляющаяся парциальными и генерализованными приступами при локально обусловленных и генерализованных формах эпилепсии соответственно.
В патогенезе локально обусловленных форм эпилепсий можно выделить четыре последовательные фазы нейроморфологических и нейрофизиологических изменений: образование эпилептогенного очага, первичного эпилептического очага, эпилептических систем, эпилептизация мозга. Эпилептогенный очаг (эпилептогенное повреждение) – очаговое поражение головного мозга вследствие воздействия факторов приобретенной и (или) врожденной предрасположенности. Структурные и функциональные нарушения мозговой ткани приводят к формированию в зоне поражения и вокруг нее эпилептического очага – группы эпилептических нейронов, генерирующих пароксизмальные деполяризационные сдвиги и вовлекающих в очаг все новые нейронные популяции. Пока эта активность ограниченна, она не дает приступообразных клинических проявлений. На стадии образования эпилептической системы в процесс вовлекаются подкорково-стволовые структуры за счет формирования между ними и корковым эпилептическим очагом устойчивых межнейрональных связей. В результате в мозге больного эпилепсией образуются системы, по которым чрезмерные очаговые нейронные разряды из эпилептического очага распространяются и вызывают эпилептические припадки. Большая роль в образовании устойчивых патологических межнейронных связей отводится нарушению функционирования антиэпилептической системы. Способностью препятствовать распространению чрезмерных нейронных разрядов обладают мозжечок, хвостатое ядро, латеральное ядро гипоталамуса, каудальное ретикулярное ядро моста, неспецифические ядра ретикулярной формации ствола. Эпилептизация мозга формируется на фоне длительно текущего эпилептического процесса, характеризуясь выраженными функционально-деструктивными изменениями с формированием в мозге больного эпилепсией большого количества устойчивых патологических межнейрональных связей.
Патогенез развития генерализованных форм эпилепсий, при которых возникает внезапная и синхронная эпилептическая активность всех корковых нейронов, не до конца ясен. Последние экспериментальные исследования показывают, что основной патологический механизм связан с таламо-корковыми нейрональными сетями. Согласно корково-таламической концепции разряд, возникающий в любом участке данной сети, мгновенно распространяется на интраламинарные ядра таламуса, а далее через таламо-корковый путь билатерально и синхронно вовлекает оба полушария. Это возможно только при условии аномально высокой возбудимости таламо-корковой системы, которая, вероятно, генетически детерминирована и обусловлена нестабильностью мембран нейронов с невозможностью поддерживать нормальный градиент концентрации ионов Na, K, Cl, Са.
В Классификацию эпилепсии ILAE 2017 г. при распределении этиологических групп был сделан акцент на те группы, которые могут быть важны для выбора тактики течения. Это структурная, генетическая, инфекционная, метаболическая и иммунная этиология, а также неизвестная этиология. Знание структурной этиологии имеет решающее значение для выбора оперативного вмешательства, а генетической – для генетического консультирования членов семьи и выбора инновационных методов таргетной медикаментозной терапии [18].
Коморбидные состояния
Коморбидные состояния различаются по типу и тяжести, от малозаметных трудностей в обучении до выраженных расстройств интеллектуальных и психических функций, таких как расстройства аутистического спектра, депрессии и проблемы с адаптацией в социуме. В более тяжелых случаях эпилепсии может наблюдаться целыйкомплекс сопутствующих заболеваний, включая моторный дефицит, такой как детский церебральный паралич, сколиоз, инсомнию и желудочно-кишечные расстройства. При постановке диагноза пациенту с эпилепсией важно, чтобы наличию коморбидных состояний уделялось должное внимание на раннем этапе, с целью обеспечить их раннюю идентификацию, диагностику и должный контроль [18].
Изменения в терминологии и определениях
Эксперты ILAE отмечают необходимость уточнения термина «эпилептическая энцефалопатия». Он должен использоваться не только при тяжелой эпилепсии в младенческом и детском возрасте, но применительно к пациентам любого возраста с эпилепсиями любой степени тяжести, как генетической этиологии, так и иной (например, структурной этиологии, при гипоксически-ишемическом повреждении ЦНС или инсульте).Термин «возрастная энцефалопатия» может применяться в тех случаях, когда имеется нарушение развития без частых эпилептических приступов, ассоциированных с регрессом или замедлением дальнейшего развития («энцефалопатия развития»). Термин «возрастная и эпилептическая энцефалопатия» может применяться в случаях, где оба фактора играют определенную роль (часто невозможно распознать, какой из них является доминирующим). Многие пациенты с такими расстройствами классифицировались ранее как имеющие «симптоматическую генерализованную эпилепсию». Этот термин больше не будет использоваться, поскольку он применялся к крайне гетерогенной группе пациентов: у пациентов с возрастной энцефалопатией и эпилепсией (то есть статической умственной отсталостью и нетяжелой формой эпилепсии).
Эксперты ILAE произвели замену термина «доброкачественный» терминами «самокупирующийся» и «фармакореактивный», то есть дающий позитивную реакцию в ответ на фармакотерапию. Термин «самокупирующийся» означает самостоятельное разрешение эпилептического синдрома. Термин «фармакореактивный» означает, что эпилептический синдром, может контролироваться с помощью соответствующей антиэпилептической терапии. Однако не все пациенты с такими синдромами будут отвечать на терапию АЭП.
Эпидемиология
Эпидемиология:
Полноценный популяционный анализ эпидемиологии эпилепсии у детей в Кыргызской Республике не проведен. Поэтому этот раздел базируется в основном на данных зарубежных авторов. Хотя на основании изучения эпидемиологии эпилепсии и обусловленной ею инвалидности и смертности можно составить представление об ущербе обществу и прогнозе развития заболевания, возможности планирования медицинской помощи больным эпилепсией, а также определить стратегию снабжения антиэпилептическими препаратами. До конца 1970-х годов прогноз считался крайне неблагоприятным. Пессимистическое отношение к прогнозу эпилепсии было опровергнуто после применения популяционного метода, который показал, что хороший исход является скорее нормой, чем исключением. До 85% больных, в детском возрасте, достигают пятилетней ремиссии в результате применения современных антиэпилептических препаратов (AicardiJ., 1994).
Распространенность:
Эпилепсия является одним из самых распространенных расстройств нервной системы, оказывающим значительное влияние на качество жизни пациента и членов его семьи. Распространенность в развитых странах составляет 5,8 чел. на 1000 населения, в развивающихся странах – 10,3 чел. на 1000 населения в городских поселениях и 15,4 чел. на 1000 населения в сельских районах. В РФ распространенность составляет 3,2 чел. на 1000 населения (Европейская часть – 3,1; Сибирь и Дальний Восток – 3,4; крупные города – 3,1; небольшие города и сельская местность – 3,7 чел. на 1000 населения соответственно). Значение распространенности эпилепсии в популяционных исследованиях детей всех возрастов Северной Америки лежит в интервале от 3,9 до 6,0 на 1000 (HauserW.A., HaererA.F.). Распространенность в Европе колеблется между 2,8 на 1000 в Англии (Verity) и 9,2 в Турции (OkanN., 1995). Почти 80% детей страдающих эпилепсией проживают в странах с низким и средним уровнем дохода. Три четверти из них не получают должного лечения; нередко они, как и члены их семей, страдают от дискриминации.
Заболеваемость:
Исследования в развитых странах продемонстрировали, что заболеваемость эпилепсией в первый год жизни и в раннем детстве высока, стабилизируется после пубертата и в целом ниже во взрослые годы до пятого десятилетия жизни (ForsgrenL. etal, 1996; OlafssonE. etal., 2005). Заболеваемость затем увеличивается в старших возрастных группах. В развивающихся странах заболеваемость была высока в детские годы, но увеличения в старших возрастных группах не было выявлено (Tekle– HaianotR. etal., 1997). Cстандартизированная по возрасту заболеваемость эпилепсией в европейских исследованиях лежит в интервале от 26 на 100000 человек в год в Норвегии (DeGraafA.S., 1974) до 47 на 100000 человек в год в Англии (MacDonaldB.K. etal., 2000). Исследования демонстрируют увеличение заболеваемости эпилепсией в группах с низким социально-экономическим статусом, по сравнению с популяцией с большим достатком. В Великобритании результаты проспективного исследования показали, что лица, относящиеся к группе с самым низким социально-экономическим статусом были более, чем в два раза склонны к развитию эпилепсии, по сравнению с группой с наиболее высоким статусом (HeaneyD.C. etal., 2002).
Текущее состояние проблемы в Кыргызской Республике.
Распространенность Эпилепсии и эпилептических синдромов в Кыргызской Республике неизвестна. Неврологами диагноз эпилепсии у детей выставляется необоснованно часто, основываясь только на данные электроэнцефалографических исследований. Ошибки в диагностике связаны с тем, что нет возможности проведения видео-ЭЭГ-мониторирования, ключевой показатель для постановки диагноза эпилепсии у детей. Так как без этого нельзя определить тип припадка и характер приступов, и соответственно нельзя правильно поставить диагноз и назначить адекватное лечение. В Кыргызстане в последние десятилетия сложилась очень неблагоприятная социальная ситуация, обусловливающая рост заболеваемости у детей неврологическими нарушениями. Наблюдается рост рождения детей с пре- и перинатальными повреждениями нервной системы, врожденными пороками развития (ВПР), наследственными заболеваниям. Это привело к резкому росту детей с ограниченными возможностями здоровья (ОВЗ), на 01.01.2019г. 29 800 зарегистрированных детей с ОВЗ. Из этого числа 33% инвалидность связана заболеваниями нервной системы, 16% связано психическими заболеваниями и 23% составляют дети с пороками развития. Значительную часть среди них составляют дети с эпилептическими припадками. В тоже время переход системы оказания медицинской помощи на семейную медицину привел к сокращению узких специалистов. В связи с этим ухудшилось диспансерное наблюдение детей с ОВЗ и особенно наблюдение больных с эпилептическими припадками. Нет единой и функционально полноценной системы комплексной медикопсихолого-педагогической помощи детям с отклоняющимся поведением. В данном контексте разработка клинического руководства оказания клинико-психолого-психиатрической помощи данной категории выглядит исключительно уместной и своевременной.
Диагностика (амбулатория)
ДИАГНОСТИКА НА АМБУЛАТОРНОМ УРОВНЕ:
1) Диагностические критерии
Жалобы:
Собираются у свидетелей приступа (отец/мать) и родственников больного.
Жалобы на пароксизмальные состояния (приступы):
• характер приступа: с утратой сознания, без утраты сознания, судорожные, бессудорожные), частота, продолжительность, наличие ауры;
Анамнез:
• первый или повторный;
• наличие в анамнезе неонатальных и фебрильных приступов;
• наследственной отягощенности по эпилепсии;
• возраст дебюта;
• наличие токсических, гипоксически-ишемических, травматических и инфекционных поражений мозга, включая внутриутробный период;
• нарушения режима приема противосудорожных препаратов;
• нарушение режима труда и отдыха);
• постприступное состояние (описание, видеозапись, ведение дневника приступов).
Физикальное обследование:
• общесоматический статус: общее состояние и его тяжесть, дыхание, пульс, АД, температура тела, измерение массы и роста пациента, окружности головы, осмотр кожных покровов,
• неврологический статус: уровень сознания, общемозговая симптоматика, менингеальные знаки, черепные нервы, двигательно-рефлекторная сфера, чувствительная сфера, координаторная функция, функции тазовых органов, когнитивные функции, вегетативная нервная система, психоэмоциональный статус.
Лабораторные исследования:
• общий анализ крови – при длительном приеме антиэпилептических препаратов, могут возникнуть побочные действия со стороны крови: снижение количества тромбоцитов, снижение свертываемости крови;
• биохимический анализ крови (АЛТ, АСТ, тимоловая проба, билирубин общий амилаза, щелочная фосфатаза, общий белок, мочевина, креатинин (по показаниям);
• исследование мочи общеклиническое (общий анализ мочи) 2 раза в год;
• определение уровня АЭП в крови необходимо:
– низкая эффективность терапии;
– начало антиэпилептической терапии, изменение дозы, изменение торговой версии препарата, изменение сопутствующей терапии;
– оценки побочных эффектов АЭП;
– доказательства стойкого снижения комплаентости терапии;
Инструментальные исследования:
• Электроэнцефалография – рутинная, позволяет определить наличие патологической электрической активности, указывает на расположение эпилептического очага;
• Длительное ЭЭГ - мониторирование – для уточнения типа приступа и регистрации иктального ЭЭГ;
• МРТ головного мозга – для выявления органической патологии;
• КТ головного мозга – при отсутствии МРТ и при противопоказаниях;
• МРТ головного мозга (по показаниям) по эпилептологическому протоколу – метод диагностики входящий в структуру предоперационного обследования, фармокорезистентных формах эпилепсии, для диагностики мезиального темпорального склероза, сосудистых мальформаций, дисгенезий мозга, дифференцировка патологического участка между ФКД и глиальными образованиями, при метаболических, митохондриальных энцефалопатиях и при очагах неясной этиологии;
• Нейросонография;
• УЗИ органов брюшной полости.
2) Диагностический алгоритм:
Рисунок – 3.
Алгоритм действий при пароксизмальных состояниях.
Диагностика (скорая помощь)
ДИАГНОСТИКА НА ЭТАПЕ СКОРОЙ НЕОТЛОЖНОЙ ПОМОЩИ:
Диагностические мероприятия:
уровень сознания, характер и продолжительность приступа, контроль АД, частоты дыхания, пульс, температуру
Лечение (амбулатория)
ЛЕЧЕНИЕ НА АМБУЛАТОРНОМ УРОВНЕ:
Тактика ведения и основные принципы терапии эпилепсии у детей
Тактика лечения:
• купировать приступы;
• предупреждение развития эпилептического статуса;
• отбор пациентов для оперативного лечения при фармакорезистентных формах.
Немедикаментозное лечение:
1. Режим II;
2. Кетогенная диета (КД) – рацион питания, характеризующийся высоким содержанием жиров, ограниченным количеством белков и относительно низким содержанием углеводов, предназначенный для лечения детей и взрослых, страдающих эпилепсией. Соотношение питательных веществ: 4 или 3.5 части жиров к 1 части белков и углеводов вместе взятых.
Медикаментозное лечение:
Цель лечения:
• сохранение качества жизни пациента;
• контроль эпилептических приступов;
• профилактика побочных эффектов лечения.
Основные принципы терапии эпилепсии у детей
Основные принципы терапии эпилепсии у детей базируются на восьми так называемых «правилах Менкеса», сформулированных J.Н. Меnkеs (2000г.), которые необходимо учитывать при назначении АЭП в нейропедиатрии:
1. Назначение адекватной для данных типов припадков и эпилептических синдромов терапии одним из препаратов первого ряда (монотерапия); лечение начинают с небольшой дозы и постепенно увеличивают ее до прекращения припадков или появления признаков передозировки. При недостаточном клиническом эффекте лечения уточняют диагноз, проверяют регулярность приема препарата (комплаенс), а также достижение максимальной переносимой дозы. Как правило, у 70% больных верно подобранная монотерапия обеспечивает адекватный контроль припадков.
1.1. Препаратами выбора при парциальных припадках (без вторичной генерализации или вторично-генерализованных) являются вальпроаты или карбамазепины. При резистентности к карбамазепинам и вальпроатам или их плохой переносимости применяют новые АЭП (топирамат, тиагабин, ламотриджин, габапентин) в качестве дополнительной терапии или монотерапии (если таковая предусмотрена инструкцией). Из перечисленных новых препаратов наиболее сильное противоэпилептическое действие (критерий эффективности – доля больных с 50%-ным уменьшением частоты припадков) в режиме дополнительной терапии при парциальной эпилепсии имеет топирамат; наилучшая переносимость (критерий - число отказов от лечения) отмечена у габапентина.
1.2. При генерализованных припадках - первично-генерализованных тонико-клонических, абсансах (особенно в сочетании с генерализованными припадками в рамках синдромов идиопатической генерализованной эпилепсии), миоклонических - препаратами выбора являются вальпроаты; карбамазепины и фенитоин противопоказаны при абсансах и миоклонических припадках. При простых абсансах препаратами выбора являются вальпроаты или этосуксимид. Атипичные абсансы, атонические и тонические припадки зачастую оказываются резистентными к лечению. В индивидуальных случаях может быть эффективен один из следующих препаратов: фенитоин, вальпроат, ламотриджин, клоназепам, этосуксимид, фенобарбитал, ацетазоламид и глюкокортикоиды или их сочетание. При миоклонических припадках препаратом выбора является вальпроат натрия, применяют также клоназепам, ламотриджин. При недостаточной эффективности или плохой переносимости традиционных АЭП применяют новые антиконвульсанты (например, ламотриджин или топирамат).
1.3. При недифференцированных припадках следует применять препараты вальпроевой кислоты, вне зависимости от половой принадлежности и зрелости.
2. Только в случаях неэффективности правильно подобранной монотерапии (после не менее двух последовательных попыток применения препаратов в режиме монотерапии) возможно использования рациональной политерапии (речь идет о препаратах первого выбора, считающихся адекватными для конкретного типа эпилептических припадков). В ее проведении следует придерживаться определенных правил. Рациональная политерапия исходит из представлений о фармакодинамике, т.е. взаимодействия препарата с нейрональным субстратом на уровне мембран нейронов и синаптических образований, обусловливающего его специфический лечебный эффект или побочные действия.
Теоретически нецелесообразно комбинировать препараты с одним и тем же преимущественным механизмом действия, целесообразно применять препараты с взаимодополнительными свойствами.
Если полезное фармакодинамическое взаимодействие является ожидаемым, то следует иметь в виду возможность и нежелательных взаимодействий в виде потенцирования побочных эффектов (особенно седативных). Так фармакодинамическое взаимодействие вальпроатов усиливает действие нейролептиков, антидепрессантов, наркотиков и снотворных. Аналогичные побочные психические эффекты дает комбинация тиагабина и вальпроата.
Таким образом, современная политерапия, являясь вынужденной мерой, должна исходить из известных на основании клинического опыта и действительно изученных, а не предполагаемых, нейрофармакологических механизмов взаимодействия лекарств, позволяющего априорно ожидать положительного эффекта. Во избежание трудноуправляемых и труднопредсказуемых эффектов сложного взаимодействия рекомендуется ограничиваться дуотерапией и в исключительном случае - комбинацией не более трех противосудорожных препаратов. При учете этих аспектов рациональная политерапия позволяет не только более эффективно подавлять припадки, но и достигать этого на более низких дозировках препаратов и соответственно при меньших побочных эффектах и с лучшими результатами в отношении когнитивных и психических функций.
2.1. При продолжении припадков на фоне монотерапии целесообразно своевременное назначение второго препарата. При хорошем клиническом эффекте возможна отмена первого препарата. Длительное лечение двумя препаратами осуществляют исключительно при невозможности проведения адекватной монотерапии. Кроме того, возможна постепенная замена первого дополнительного препарата (в случае его неэффективности) другим дополнительным препаратом.
2.2 Лечение тремя препаратами целесообразно только при неэффективности терапии двумя адекватными препаратами.
3. Частота приема АЭП определяется их временем полувыведения. Во всех случаях следует стремиться к минимально допустимой кратности приема конкретных препаратов (не более 2 раз в день). По современным представлениям, весьма целесообразным выглядит использование пролонгированных (так называемых «ретардных») форм АЭП, например депакина-хроно, финлепсина ретарда или тегретола ЦР. Хотя практическое использование пролонгированных форм антиконвульсантов в детской неврологии обычно сопряжено с рядом возрастных ограничений, в целом отмечается тенденция к их широкому и преимущественному применению. Количество побочных эффектов при назначении детям пролонгированных форм АЭП существенно уменьшается по сравнению с использованием их обычных (традиционных) форм. Имеющиеся данные подтверждают не только лучшую переносимость, но и более высокую клиническую эффективность АЭП пролонгированного действия, что объясняется преимущественно достижением стабильной концентрации препаратов в плазме крови пациентов. Время приема антиконвульсантов определяется не только особенностями эпилептического синдрома (временем возникновения и особенностями развития приступов и т.д.), но и собственными характеристиками препаратов (побочные эффекты и т.п.). Так, например, фенобарбитал (и его производные), обладающие сравнительно длительным временем полувыведения, а также выраженным седативным эффектом, можно назначать однократно, в вечерние часы. Тем не менее, более предпочтительным является двукратный прием фенобарбитала, позволяющий избежать резких колебаний концентрации препарата в крови. Некоторые АЭП, особенно назначенные в высоких дозах, необходимо принимать 3 раза вдень во избежание развития побочных эффектов. Известно, что у детей препараты метаболизируются быстрее, чем у взрослых. Поэтому при назначении лечения пациентам педиатрического возраста целесообразным является более частый прием АЭП и использование более высоких доз (в расчете на 1кг массы тела).
4. Отмена АЭП должна быть постепенной, с обязательным учетом формы эпилепсии и ее прогноза, возможности возобновления припадков, индивидуальных и возрастных особенностей пациента (во внимание следует принимать как медицинские, так и социальные факторы). Отмену противоэпилептической терапии проводят, как правило, не менее чем через 2-3 года после полного прекращения припадков (рекомендуют также и срок до 5 лет), под контролем данных ЭЭГ- исследования.
5. Терапия должна быть «корректной» - необходим учет интересов больного и экономических аспектов лечения (баланс эффективности, побочных эффектов и стоимости препаратов).
6. Фармакорезистентность - продолжение припадков, несмотря на адекватное противоэпилептическое лечение, включая комбинированную терапию (минимум двумя препаратами, содержание которых в плазме соответствует или превышает необходимый терапевтический уровень), требует дополнительного обследования больного и решения вопроса о хирургическом лечении. Первичной целью хирургических методов лечения эпилепсии, является уменьшение выраженности эпилепсии и/или урежение эпилептических приступов. Другие альтернативные методы лечения эпилепсии у детей. Кетогенные диеты (КД) на сегодняшний день рассматриваются в качестве эффективного экспериментального лечения эпилептических синдромов
7. Определение концентрации АЭП в крови является одним из основных требований Международных стандартов ведения больных эпилепсией, приобретая особое значение при лечении пациентов педиатрического возраста. Мониторинг концентрации антиконвульсантов в крови считается необходимым при использовании фенитоина (дифенина), карбамазепинов, препаратов вальпроевой кислоты, фенобарбитала, этосуксимида, а также примидона. В фармакотерапии эпилепсии указанный мониторинг приобрел столь большое значение в связи с тем, что для большинства упомянутых выше АЭП были выявлены статистически значимые корреляции между терапевтической эффективностью лекарственных средств и их крови обследуемых пациентов.
8. Лечение эпилепсии у детей (и подростков) требует знания особенностей фармакокинетики и фармакодинамики АЭП у этого вариабельного в возрастном отношении контингента (от рождения до 18 лет). В детской неврологии, наряду с чисто медицинскими проблемами, особую значимость приобретают контакт и сотрудничество с родителями пациента (и другими членами его семьи), разъяснение целей проводимого лечения, необходимости соблюдения регулярного приема препаратов, а также возможных проявлений побочных реакций. Трудности фармакотерапии эпилептических синдромов у пациентов педиатрического возраста сопряжены с этиологией эпилепсии, наличием сопутствующей патологии, возможностью взаимодействия АЭП с другими средствами, принимаемыми детьми для лечения соматических состояний, а также с возрастными особенностями абсорбции и метаболизма лекарственных препаратов. Выбор АЭП для детей определяется классификационной принадлежностью имеющихся припадков, анамнезом болезни, а также данными ЭЭГ- исследования. В детской неврологии основной целью является использование в контроле за припадками только одного (эффективного) антиконвульсанта, пригодного к использованию с учетом возраста пациента, а также обладающего наименьшим числом побочных эффектов. Имеются данные, подтверждающие целесообразность более широкого применения в педиатрической практике некоторых новых АЭП (в частности, ламотриджина и топирамата).
Не подлежит сомнению, что лечение эпилепсии не должно ограничиваться сугубо медицинскими мероприятиями, а включать и ряд социальных мер. В современной неврологии (и эпилептологии) одной из приоритетных целей является улучшение качества жизни и реабилитации пациентов, страдающих эпилепсией.
Пациенты педиатрического возраста нуждаются в реабилитации в большей мере, поскольку детский возраст характеризуется отсутствием социальной зрелости и независимости.
Таблица 12 4.3.
Варианты лечения инфантильных спазмов
|
Терапия
|
Доза
|
Класс
|
N
|
Изучение
|
Этиология
|
Эффективность в прекращении клинических спазмов
|
Эффективность при прекращении клинических спазмов и ликвидации гипсарритмии
|
Эффективность в нормализации когнитивного исхода
|
|
АКТГ (высокая доза)
|
150 Ед / м2 / д × 2 недели, затем постепенная отмена
|
I/II
|
15
|
Baram 1996
|
Комбинированный
|
-
|
87% (через 2 недели)
|
-
|
|
|
150 Ед / м2 / д × 3 недели, затем постепенная отмена
|
I/II
|
26
|
Hrach ovy, 1994
|
Комбинированный
|
-
|
50% (в течение 3 мес.)
|
-
|
|
|
Тетракозактид 1 мг (эквивалент АКТГ 100 Ед / сут) к-ые 48 ч × 2 недели, затем постепенная отмена
|
III
|
37
|
Kivity , 2004
|
Комбинированный
|
-
|
100% (лечение в течение 1 месяца), 87% (лечение через 1 месяц), оценка через 1–4 недели
|
100% (лечение в течение 1 месяца), 47% (лечение через 1 месяц), оценка через 6–21 год
|
|
|
110 Ед / м2 / д × 3 недели, затем постепенная отмена
|
III
|
12
8 |
Lombr osso, 1983
|
Комбинированный
|
48% (через 10 мес.)
|
43% (через 10 мес.)
|
50% (AКТГ 8 недель), 59% (AКТГ 8 недель + 14 недель оральных стероидов) через 6 лет (в криптогенных случаях n = 73)
|
|
|
Тетракозактид 0,75 мг (эквивалент AКТГ 75 Ед / сут) кые 48 ч × 2 недели, затем постепенная отмена
|
I/III
|
25
|
Lux, 2004
|
Комбинированный
|
76% (через 2 недели)
|
-
|
-
|
|
АКТГ (низкая доза)
|
20–30 ед / д, затем постепенная отмена
|
I/II
|
24
|
Hrach ovy, 1994
|
Комбинированный
|
-
|
58% (в течение 6 недель)
|
-
|
|
Оральный стероиды
|
Преднизон 2 мг / кг / сут × 2 недели, затем постепенная отмена
|
I/II
|
14
|
Baram , 1996
|
Комбинированный
|
-
|
29% (в течении 2 недель)
|
-
|
|
|
Преднизолон 40–60 мг / сут × 2 недели, затем постепенная отмена
|
I/III
|
30
|
Lux, 2004
|
Комбинированный
|
70% (в течении 2 недель)
|
-
|
-
|
|
|
Преднизолон 8 мг / кг / сут (максимум 60 мг / сут) × 2 недели
|
III
|
27
|
Hussai n, 201
|
Комбинированный
|
-
|
63% (в течении 2 недель)
|
-
|
|
|
Преднизолон 2 мг / кг / сут × 8 недель, затем постепенная отмена
|
III
|
77
|
Lombr osso, 1983
|
Комбинированный
|
38% (через 10 месяцев)
|
35% (через 10 месяцев)
|
12% через 6 лет (в криптогенных случаях n = 17
|
|
Гормональная терапия
|
Преднизолон 40–60 мг / сут или тетракозактид 0,75 мг каждые 48 ч × 2 недели, затем постепенная отмена
|
I/III
|
55
|
Lux, 2005
|
Комбинированный
|
75% (в 12– 14 мес)
|
-
|
в криптогенной группе ШАПV 88,2 (лучше, чем в группе с вигабатрином 78,9) в 12–14 мес.
|
|
Вигабатрин
|
Начальная доза 50 мг / кг / сут, максимум 150 мг / кг / сут
|
I
|
20
|
Аpplet on, 1999
|
Комбинированный
|
Комбинированные 35% (VGB) против 10% (плацебо) через 5 дней, 42% после 24 недель с открытой фазой
|
25% (VGB) против 5% (плацебо) через 5 дней
|
-
|
|
|
Началь ная доза 50 мг / кг / сут, максимальная 150 мг / кг / сут.
|
I/III
|
52
|
Lux, 2004
|
Комбинированный
|
52% (через 2 недели)
|
|
|
|
|
Началь ная доза 50 мг / кг / сут, максимальная 150 мг / кг / сут.
|
I/III
|
52
|
Lux, 2005
|
Комбинированный
|
76% (в 12– 14 мес)
|
-
|
в криптогенной группе ШАПV 78,9 (хуже, чем в группе гормональной терапии 88,2) в 12–14 мес.
|
|
|
Началь ная доза 50 мг / кг / сут, максимальная 150 мг / кг / сут.
|
IV
|
18
0 |
Djuric , 2014
|
Комбинированный
|
-
|
56% (через 2 недели)
|
44% (средний период наблюдения, 11 лет после лечения)
|
|
Вальпроат
|
Началь ная доза 15–20 мг / кг / сут, максимальная 60 мг / кг / сут.
|
IV
|
19
|
Bach man, 1982
|
Комбинированный
|
42% (в течение 2 лет)
|
-
|
-
|
|
Топиромат
|
Началь ная доза 25 мг / сут, максимальная 24 мг / кг / сут.
|
IV
|
11
|
Glaus er, 1998
|
Комбинированный
|
-
|
45% (в течение 90 дней)
|
-
|
|
Зонисамид
|
Началь ная доза 3- 5 мг / кг / сут, максимум 10 мг / кг / сут.
|
IV
|
11
|
Suzuk i, 1997
|
Комбинированный
|
-
|
36% (через 3 недели)
|
-
|
|
|
4-13 мг / кг / сут.
|
IV
|
|
Suzuk i, 2002
|
Комбинированный
|
-
|
20% (через 3 недели)
|
У 5/7 (71%) соответствующих был нормальный КФ (средний период наблюдения, через 53 месяца после лечения)
|
|
Леветирацетам
|
Началь ная доза 20 мг / кг / сут, максимальная 80 мг / кг / сут.
|
IV
|
7
|
Mikati , 2008
|
Комбинированный
|
-
|
14%
|
-
|
|
Пиридоксин
|
Пиридоксальфосфат 30–400 мг / сут.
|
IV
|
11
8 |
Ohtsu ka, 1987
|
Комбинированный
|
13%
|
13%
|
43% соответствующих
|
|
Кетогенная диета
|
-
|
II
|
13
|
Kosso ff, 2008
|
Комбинированный
|
62% (в 1 мес.)
|
9% (в 1 мес.)
|
-
|
|
Операция
|
-
|
IV
|
65
|
Chuga ni, 2015
|
пораженные случаи
|
71% класс I (средний период наблюдения 45 месяцев)
|
-
|
-
|
|
|
|
IV
|
47
|
Shield s, 2002
|
пораженные случаи
|
60% класс I (в 24–60 мес)
|
-
|
-
|
Сокращения: АКТГ, адренокортикотропный гормон; КФ, Коэффициент развития; ШАПV, шкалы адаптивного поведения Vineland; VGB, вигабатрин.
Рис. 5. Рекомендуемое руководство по лечению в странах с низким и средним уровнем дохода на основе существующих данных.
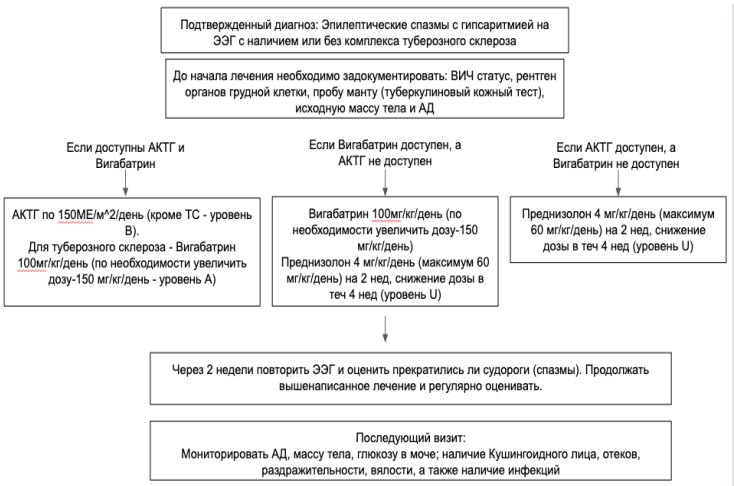
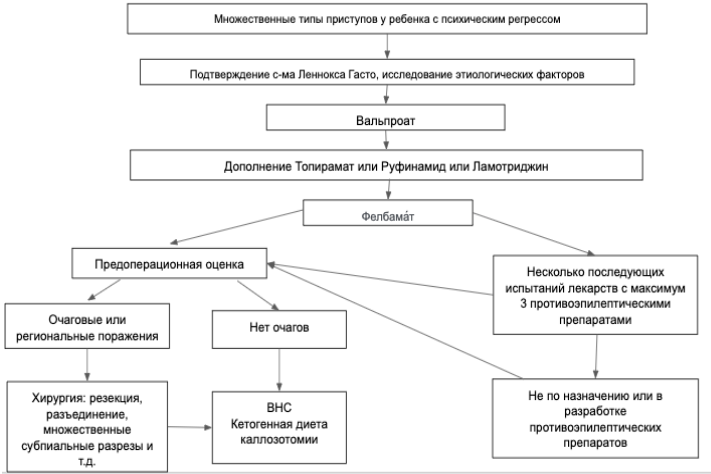
Контроль эффективности лечения:
1. Клинический – ведение дневника приступов!
2. Контроль за приступами и приемом препаратов у детей, родителями
3. Нейрофизиологический (ЭЭГ, видео-ЭЭГ-мониторинг)
4. Терапевтический лекарственный мониторинг
5. Лабораторно-биохимический мониторинг
6. Ревизия
7. Прогноз
Далее есть повторы
Препараты первого и второго выбора в лечении различных форм эпилепсий и типов эпилептических приступов, а также препараты, назначения которых следует избегать из-за риска усиления приступов (адаптировано из NICE, 2004.), представлены в таблице.
Таблица 13
Препараты первого и второго выбора в лечении различных форм эпилепсий и типов эпилептических приступов
|
Тип приступов/ синдром
|
Первого выбора
|
Второго выбора
|
Избегать
|
|
Фокальные приступы
|
CBZ, LTG, OXC, TPM, VPA
|
CLB, GBP, LEV, PHT, TGB
|
|
|
Генерализованные приступы:
|
|
|
|
|
Абсансы
|
ESM, LTG, VPA
|
CLB, CLN, TPM
|
CBZ, GBP, OXC, TGB, VGB
|
|
ГТКП
|
CBZ, LTG, VPA, TPM
|
CLB, LEV, OXC
|
TGB, VGB
|
|
Миоклонические
|
VPA, TPM
|
LTG, CLB, CLN, LEV, piracetam
|
CBZ, GBP, OXC, TGB, VGB
|
|
Тонические
|
LTG, VPA
|
CLB, CLN, LEV
|
CBZ, OXC
|
|
Атонические
|
LTG, VPA
|
TPM CLB, CLN, LEV, TPM
|
CBZ, OXC, PHT
|
|
Детская абсансная эпилепсия
|
ESM, LTG, VPA
|
LEV, TOP
|
CBZ, OXC, PHT, TGB, VGB
|
|
Юношеская абсансная эпилепсия
|
LTG, VPA
|
LEV, TPM
|
CBZ, OXC, PHT, TGB, VGB
|
|
Юношеская миоклоническая эпилепсия
|
LTG, VPA
|
CLB, CLN, TPM, LEV
|
CBZ, OXC, PHT, TGB, VGB
|
|
BECTS
|
CBZ, LTG, OXC, VPA
|
LEV, TPM
|
|
|
BCOS
|
CBZ, LTG, OXC, VPA
|
LEV, TPM
|
|
|
Инфантильные спазмы
|
VGB, стероиды
|
CLB, CLN, VPA, TPM
|
CBZ, OXC
|
|
Синдром Драве
|
VPA, TPM, CLB, CLN
|
LEV, стирипентол
|
CBZ, LTG, OXC, VGB
|
|
Синдром Леннокса ‒ Гасто
|
VPA, LTG, TPM
|
CLB, CLN, ESM, LEV
|
CBZ, OXC
|
|
Синдром Ландау ‒ Клеффнера
|
Стероиды, LTG, VPA
|
LEV, TPM
|
CBZ, OXC
|
|
CSWS
|
Стероиды, CLB, CLN, VPA, LTG, ESM,
|
LEV, TPM
|
CBZ, OXC, VGB
|
|
Миоклоническая астатическая эпилепсия
|
CLB, CLN, VPA, TPM
|
LEV, LTG
|
CBZ, OXC
|
Примечание:
CBZ ‒ carbamazepine; CLB ‒ clobazam; CLN ‒ clonazepam; ESM ‒ ethosuximide; GBP ‒ gabapentin; LEV ‒ levetiracetam; LTG ‒ lamotrigine; OXC ‒ oxcarbazepine; PHT ‒ phenytoin; VPA ‒ sodium valproate; TGB ‒ tiagabine; TPM ‒ topiramate; VGB ‒ vigabatri
Эпилептический статус: определение и основные принципы лечения
В 1981 г. ILAE определяла ЭС как приступ, сохраняющийся в течение достаточно продолжительного времени или как отдельные приступы, повторяющиеся достаточно часто, когда восстановление сознания между приступами не происходит. Следующее определение было дано в 1989 г.: эпилептический статус определяется как длительный (более 30 мин) эпилептический приступ или частые приступы, следующие друг за другом без полного восстановления сознания.
Основополагающим показателем следует считать не фактор времени, а тяжесть состояния пациента. Если у больного было всего лишь несколько припадков и он находится в сопоре, а тем более в коме, это состояние следует расценивать как эпилептический статус. (Карлов В.А., 2003).
Согласно определению ILAE 2015 г. эпилептический статус – это состояние, являющееся результатом либо неспособности механизмов, ответственных за прекращение эпилептических приступов, осуществлять свою функцию, либо появление механизмов, приводящих к аномально длительным приступам (после момента времени t1). Также ЭС – это состояние, которое может иметь долгосрочные последствия (после момента времени t2), включающие гибель нейронов, повреждение нейронов и изменение нейронных сетей в зависимости от типа и продолжительности приступов. Для тонико-клонического ЭС время t1 составляет 5 мин, t2 – 30 мин. Для фокального ЭС с нарушением сознания время t1 составляет 10 минут, t2 – более 60 мин. Для ЭС абсансов время t1 составляет 10–15 мин, t2 – неизвестно. Относительно ЭС абсансов в дальнейшем планируется уточнение времени t1 и t2.
К эпилептическому статусу может приводить несоблюдение пациентом режима, резкое снижение дозы АЭП, замена или отмена препаратов, переход с оригинальных препаратов на генерики, назначение неадекватной терапии, инфекционные заболевания с лихорадкой, травмы. Смертность при ЭС в условиях отсутствия специализированной помощи составляет до 50 %, в то время как при адекватном лечении ‒ 5‒12 % (Мухин К.Ю., 2008).
Выделяют следующие стадии эпилептического статуса: предстатус (продолжительность приступа до 9 мин), начальный ЭС (10‒30 мин), развернутый ЭС (31‒60 мин) и рефрактерный ЭС (свыше 60 мин).
Лечебные мероприятия дифференцированы в зависимости от стадии эпилептического статуса (Мухин К.Ю., 2008):
1. Предстатус
-
Обеспечение проходимости дыхательных путей.
-
Оксигенотерапия.
-
Диазепам (1 ампула 2 мл ‒ 10 мг) ректально или в/в медленно в разовой дозе 0,25 мг/кг. Скорость в/в введения диазепама не более 2 мг/мин. Суммарная доза препарата в сутки не должна превышать 5 мг у детей до 5 лет; 20 мг для детей 6‒12 лет и 40 мг для детей старшего возраста. Предпочтительнее использовать лоразепам или мидазолам.
-
При отсутствии эффекта ‒ вальпроат для инъекций (1 ампула 4 мл ‒ 400 мг). Препарат вводится в/в медленно в разовой дозе 15 мг/кг (в домашних условиях родители могут вводить препарат внутримышено). Можно повторно вводить через 12 часов до 2000 мг в сутки.
2. Ранний статус
-
Диазепам в/в (см. предстатус).
-
При отсутствии эффекта ‒ лоразепам (в 1 мл 4 мг) в дозе 0,1 мг/кг в/в медленно со скоростью 2 мг/мин. Вводится 1 или 2 раза с интервалом не менее 20 мин, суммарно не более 4 мг в сутки для детей. Лоразепам имеет большую продолжительность действия после однократной инъекции, чем диазепам. Побочные эффекты: развитие толерантности после 1‒2 инъекций; редко ‒ угнетение дыхания, артериальная гипотензия.
-
При отсутствии эффекта ‒ вальпроат для инъекций в/в (см. предстатус).
-
При отсутствии эффекта ‒ фенитоин для инъекций (дифантоин) (5 мл ‒ 250 мг) в/в медленно. Развести в физиологическом растворе. Дозировка 20 мг/кг со скоростью 50 мг/мин. Побочные эффекты: остановка сердца, артериальная гипотония, флебосклероз. В Европе базовым препаратом является фосфенитоин.
3. Развернутый статус
-
Диазепам или лоразепам (см. выше).
-
При неэффективности ‒ вальпроат для инъекций. Препарат вводится в/в струйно медленно в дозе 15 мг/кг. Рекомендуется введение не более трех раз в день с 8-часовым интервалом до суточной дозы не более 2000г. Возможно в/в капельное введение препарата со средней скоростью 1 мг/кг/ч. Такой режим дозирования депакина рекомендуется в течение не более трех суток, с последующим переходом на пероральный прием. Побочное действие: крайне редко развитие токсического гепатита.
-
При неэффективности ‒ фенобарбитал (1 мл ‒ 200 мг). Вводится в/в медленно. Детям до одного года 20 мг/кг, после года ‒ 10‒15 мг/кг со скоростью до 100 мг/мин. Разовая доза у детей старшего возраста не должна превышать 1000 мг. Побочные эффекты: снижение сократительной способности миокарда, угнетение дыхания, угнетение сознания, артериальная гипотензия.
4. Рефрактерный статус
-
Интубирование пациента с переводом на искусственную вентиляцию легких в реанимационном отделении.
-
Барбитуровый наркоз: введение тиопентала ( 2,5 % р-р, 1мл ‒ 25 мг) в/в очень медленно в средней дозировке 100‒250 мг для детей старшего возраста. При отсутствии эффекта дополнительное введение препарата в дозе 50 мг внутривенно каждые 3 мин до полного купирования приступов. Далее переход на поддерживающую дозу, в среднем 3‒5 мг/кг в/в каждый час (желательно с контролем уровня препарата в крови). Продолжительность барбитурового наркоза не должна превышать 12‒24 ч. Побочные эффекты: снижение сократительной способности миокарда, угнетение дыхания, артериальная гипотензия, токсический гепатит и панкреатит, анафилактический шок
-
После ликвидации ЭС и при восстановлении сознания переход на пероральный прием ПЭП.
Во время 2‒4-й стадий ЭС проводится дополнительная терапия, направленная на коррекцию жизненно важных функций, электролитных нарушений, борьбу с отеком мозга (дексаметазон 4 мг внутривенно каждые 6 ч или маннитол 1,0 г/кг внутривенно капельно со скоростью 60‒80 капель в минуту).
1. Специализированным отделениям необходимо иметь протокол терапии судорожного ЭС. Протокол должен быть согласован со смежными специалистами и может быть использован в стационарах другого профиля. При инфузионной терапии ЭС следует проводить мониторинг кардиореспираторных функций.
2. Протокол должен быть четко структурирован по времени (таблица 13).
Таблица №14
Протокол лечения ЭС ГТКП на госпитальном этапе
|
Стадия
|
Препарат
|
Примечание
|
|
1-я стадия раннего статуса (0‒10‒30 мин)
|
Лоразепам: 4 мг в/в болюсно (при необходимости ‒ повторное введение)
|
В случае продолжения судорожного синдрома более 30 мин
|
|
2-я стадия развернутого статуса (10/30‒60/90 мин)
|
Фенобарбитал: в/в инфузия 10 мг/кг, максимальная скорость введения ‒ 100 мг/мин; или фенитоин: в/в инфузия 15 мг/кг максимальная скорость введения ‒ 50 мг/мин; или фосфенитоин (РЕ): в/в инфузия 15 РЕ/кг* при скорости введения 100 мг РЕ/мин; или вальпроат: в/в инфузия 25 мг/кг при скорости введения 3‒6 мг/кг/мин
|
Если судорожный синдром продолжается более 60‒90 мин
|
|
3-я стадия резистентого ЭС (> 60/90 мин)
|
Пропофол: 2 мг/кг в/в болюсно, затем непрерывная инфузия, начиная с 5‒10 мг/кг/ч с постепенным снижением дозы, под контролем ЭЭГ-паттерна «вспышка ‒ подавление» (обычно 1‒3 мг/кг/ч); или тиопентал: 100‒250 мг в/в болюсно в течении 20 с, затем 50 мг болюсно до достижения контроля над судорогами, с дальнейшей в/в инфузией со снижением дозы, Под контролем ЭЭГ-паттерна «вспышка ‒ подавление» (обычно 3‒5 мг/кг/ч); или мидазолам: в/в инфузия 0,1‒0,3 мг/кг со скростью, не превышающей 4 мг/мин, затем непрерывная в/в инфузия со снижением дозы, под контролем ЭЭГ-паттерна «вспышка ‒ подавление» (обычно 0,05‒0,4 мг/кг/ч)
|
Если впервые 12 ч удалось установить контроль над судорожным синдромом, дозу препарата необходимо медленно снижать на протяжении 12 ч, с последующей постепенной отменой. Этот цикл может повторяться каждые 24 ч до достижения контроля над судорожным синдромом
|
3. Необходимо проведение полного комплекса неотложных мероприятий. Параллельно с проведением специфической противосудорожной терапии должны в полном объеме проводиться рутинные методы исследования.
4. Исследования, необходимые для выяснения причин развития ЭС, необходимо проводить без задержки инициальной терапии.
5. Инфузионную противосудорожную терапию предпочтительно проводить на госпитальном этапе в связи с наличием оборудования для реанимационных мероприятий и крайне нежелательно на догоспитальном. В последнем случае используются альтернативные формы противосудорожных препаратов, такие как интраназальный или буккальный мидазолам, ректальная форма диазепама.
Таблица №15
Препараты, используемые для терапии раннего ГТКП ЭС
|
Препарат
|
Форма назначения
|
Дозировка для детей
|
|
Диазепам
|
Внутривенно болюсно (≤ 2‒5 мг/мин)
Ректальная форма |
0,25‒0,5 мг/кг
0,5‒0,75 мг/кг* |
|
Клоназепам
|
Внутривенно болюсно (≤ 2 мг/мин)
|
250‒500 мг
|
|
Лоразепам
|
Внутривенно болюсно
|
0,1 мг/кг
|
|
Мидозалам
|
Буккально или интраназальные формы
|
0,15‒0,3 мг/кг*
|
7. Если терапия бензодиазепинами не дает контроля над судорожным синдромом, следует сделать вывод о наступлении 2-й стадии ЭС, что требует немедленного проведения инфузионной противосудорожной терапии. При этом наиболее часто используются фенитоин или фенобарбитал, однако существуют и альтернативные препараты (таблица 15).
8. Если лечение в стадии развернутого ЭС является неэффективным, необходимо констатировать переход в 3-ю стадию – стадию резистентного ЭС с немедленным назначением внутривенного наркоза. Он не проводится при альтернативном лечении в стадии развернутого ЭС и при отсутствии необходимых условий и ресурсов. Пациентам в стадии резистентного ЭС, которым проводится внутривенный наркоз, необходим ЭЭГ-мониторинг.
Таблица №16
Препараты, используемые для терапии развернутого тонико-клонического ЭС
|
Препарат
|
Форма назначения
|
Дозировка для детей
|
|
Фосфенитоин
|
Внутривенно болюсно (≤ 100 мг РЕ/мин)
|
Не используется
|
|
Леветирацетам
|
Внутривенно болюсно
|
Не используется
|
|
|
Внутривенно болюсно/инфузионно (≤ 50 мг/мин)
|
20 мг/ кг, 25 мг/мин
|
|
|
Внутривенно болюсно/инфузионно (≤ 100 мг/мин)
|
15‒20 мг/кг
|
|
Вальпроат
|
Внутривенно болюсно
|
20‒40 мг/кг
|
9. В случае назначения парентеральной противосудорожной терапии пациентам во всех стадиях ЭС должен проводиться тщательный контроль кардиореспираторных функций.
10.В случае ЭС, развившемся вследствие отмены или сокращения дозы противосудорожного препарата у пациентов, страдающих эпилепсией, препарат необходимо повторно назначить в инфузионной форме (при возможности).
Принципы противосудорожной терапии:
1. Догоспитальный этап: рекомендовано проведение неинфузионной противосудорожной терапии. Наиболее часто используемые препараты – интраназальный или буккальный мизадолам, ректальная форма диазепама (отмечена высокая эффективность использования буккального мидазолама в сравнении с ректальной формой диазепама). На госпитальном этапе при наличии контроля кардиореспираторных функций и необходимого оборудования для реанимационных мероприятий возможно проведение инфузионной противосудорожной терапии.
2. На госпитальном этапе для инфузионной противосудорожной терапии рекомендовано использование диазепама, клоназепама и лоразепама. По данным ряда исследований, наиболее эффективным является лоразепам.
3. Стадия развернутого ЭС: традиционная терапия в этой стадии – инфузионное введение фенитоина или фенобарбитала (внутривенное введение вальпроата натрия или левитирацетама может быть хорошей альтернативой).
4. Стадия резистентого ЭС: в отделении интенсивной терапии (ОИТ) проводится внутривенный наркоз, желательно с выполнением повторной ЭЭГ или ЭЭГ-видеомониторинга. Варианты терапии при проведении внутривенного наркоза для лечения резистентного ЭС:
-
наиболее часто используемыми препаратами являются мидазолам, пропофол или тиопентал (фенобарбитал), однако сравнительные рандомизированные исследования, способные оценить эффективность данных препаратов, не проводились. Реже используются средства для ингаляционного наркоза и кетамин;
-
проведение гипотермии на данной стадии может быть эффективным. При наличии соответствующего оборудования в ОИТ этот метод также может использоваться.
Абсансный ЭС при идиопатической генерализованной эпилепсии. При развитии абсансного ЭС неотложная терапия может проводиться на догоспитальном этапе. Принципы терапии включают использование оральных форм бензодиазепинов – лоразепама, клобазама, диазепама или мидазолама, которые являются препаратами «первой линии» и должны быть применены как можно быстрее (если необходимо – на до госпитальном этапе). В случае неэффективности данных препаратов необходимо переходить к инфузионной терапии, обеспечив госпитализацию пациента и проведение ЭЭГ-контроля. Инициальная инфузионная терапия проводится обычно невысокими дозами диазепама или лоразепама. При неэффективности терапии бензодиазепинами проводится инфузионная терапия вальпроатом натрия или леветирацетамом.
В настоящее время существует недостаточное количество достоверных свидетельств о преимуществах или недостатках тех или иных терапевтических подходов, в педиатрической практике. Безопасное использование ряда препаратов (вальпроаты, фенитоин) является, по меньшей мере, сомнительным. В острой стадии большинство противосудорожных препаратов являются эффективными. В случае неполного контроля над фокальными проявлениями они могут предотвращать вторичную генерализацию.
В хронической стадии достигнуть контроля не удается даже при введении высоких доз противоэпилептических препаратов, однако они также могут препятствовать вторичной генерализации. В некоторых случаях применяются стероиды, однако их эффективность не доказана. В данном случае противосудорожный эффект реализуется за счет их противовоспалительного действия. Использование иммуноглобулина и плазмафереза может оказывать позитивное действие также за счет вторичных влияний. Согласованные протоколы лечения EPС (доза, этапность, продолжительность) в настоящее время не разработаны.
Понятие фармакорезистентной эпилепсии
Эпилепсия может считаться фармакорезистентной, если невозможно достичь отсутствия припадков после назначения двух хорошо переносимых схем ПЭП установленного образца (моно- или политерапия) (Kwan et al., 2010). Ситуации, в которых может наблюдаться недостаточный терапевтический ответ: неверная диагностика эпилепсии, выбор не подходящего для эпилептического синдрома АЭП, отказ от приема назначенных АЭП, новообразование головного мозга, метаболические нарушения, аутоиммунный процесс, прием конкурирующего лекарственного препарата или алкоголя.
Паттерны фармакорезистентности по Kwan P., Brodie M.J.:
-
Резистентность de nova: пациент никогда не достигает ремиссии, начиная с дебюта заболевания.
-
Прогрессирующая резистентность: вначале удается достичь ремиссии, но в дальнейшем приступы возобновляются и носят неконтролируемый характер.
-
«Волнообразный паттерн» резистентности: чередование периодов с хорошим контролем и отсутствием контроля над приступами.
-
Исходная эпилепсия, резистентная к лечению, но в дальнейшем достигается контроль над приступами.
В резистентных случаях применяются хирургическое вмешательство, стимуляция блуждающего нерва, кетогенная диета. По мнению Engel (1996), Holthausen (2002), следующие эпилептические синдромы являются прямым показанием к оперативному лечению:
-
симптоматические фокальные формы эпилепсии при прогрессирующих объемных образованиях головного мозга;
-
энцефалит Расмуссена;
-
гемимегалэнцефалия;
-
эпилепсия при синдроме Штурге ‒ Вебера;
-
катастрофические фокальные формы эпилепсии в младенчестве, обусловленные кортикальными дисплазиями, с наличием полиморфных приступов, включая тонические падения, с мультирегиональными изменениями на ЭЭГ.
Фенобарбитал и другие барбитураты могут обострять абсансные приступы, особенно у пациентов с генерализованными идиопатическими эпилепсиями.
У этосуксимида показана потенциальная способность усиливать тонико-клонические приступы у детей с абсансной эпилепсией, но нет четких свидетельств этого, и оценка данных усложняется тем, что появление тонико-клонических приступов у пациентов может быть частично связано с их основным заболеванием.
Габапентин усугубляет миоклонии у пациентов с парциальной или генерализованной эпилепсией, включая больных, у которых никогда раньше не было миоклонии.
Ламотриджин усугубляет тяжелую миоклоническую эпилепсию у детей. Хотя ламотриджин успешно применялся у пациентов с ювенильной миоклонической эпилепсией, у некоторых больных из этой группы были приступы (особенно миоклонические подергивания), ухудшившиеся на фоне приема ламотриджина.
Тиагабин может провоцировать бессудорожный эпилептический статус у пациентов с генерализованными эпилепсиями и реже у пациентов с парциальной эпилепсией. Усугубление приступов вальпроатами ‒ чаще манифестация интоксикации в контексте лекарственной энцефалопатии, иногда связанной с гипераммониемией, или ранний признак угрожающей гепатотоксичности. Вигабатрин может усугублять абсансные, тонические и миоклонические приступы.
Если неэффективны монотерапии, то применяется рациональная политерапия: применение нескольких антиконвульсантов (чаще всего двух) с учетом их фармакодинамических взаимодействий. Считается, что в основу политерапии должны быть положены следующие рациональные принципы (Белоусова Е.Д., 2011):
1. Преимуществами обладают комбинации антиконвульсантов с различными механизмами действия.
2. Доза первого из назначенных препаратов должна быть адаптирована с учетом возможных лекарственных взаимодействий при комбинации со вторым препаратом.
3. Взаимодействия ферментоиндуцирующих препаратов (карбамазепин, фенитоин, фенобарбитал) с точки зрения влияния на концентрацию в крови в значительной степени непредсказуемы.
4. Следует избегать сочетания антиконвульсантов с выраженным седативным эффектом.
Вальпроат + карбамазепин:
- карбамазепин снижает концентрацию вапьпроата в крови, а вальпроат увеличивает промежуточные метаболиты карбамазепина, при этом повышает токсичность;
- нельзя назначать высокие дозы, может усиливать токсичекие дозы карбамазепина при нормальной концентрации препарата в крови;
- побочные эффекты преимущественно обусловлены карбамазепином, (атаксия, сонливость, диплопия).
Вальпроат + леветирацетам:
- синергизм по противосудорожному эффекту;
- нет фармокинетического взаимодействия, отсутствует нейротоксичность;
- назначается у детей с генерализованными, фокальными и неклассифицированными типами эпилепсии у детей;
- леветирацетам (Кеппра) обладает широким спектром действия с минимальным лекарственным взаимодействием и является препаратом первой линии для многих типов припадков у детей из-за высокого уровня безопасности.
- возможны побочные эффекты - возбуждение, изменение поведения, эффект аггравации у детей с ментальными нарушениями.
Вальпроат + ламотриджин:
- синергизм по противосудорожному эффекту;
- титрование ламотриджина в данной комбинации должно быть очень медленным, начальная доза ЛТЖ 0,2 мг/кг в сутки с повышением дозы один раз в 2 недели на 0,3 мг на кг веса в сутки, максимальная доза ЛТЖ не должна превышать 5 мг/кг в сутки;
- ЛТЖ увеличивает концентрацию вальпроата в крови;
- серьёзный побочный эффект, появление аллергической сыпи до тяжелых форм.
Вальпроат + топирамат:
- данных по синергизму не выявлено;
- фармокинетическое взаимодействие, воздействуют на разные типы эпилептических приступов;
- начальная доза топирамата 1-2 мг/кг в сутки с постепенным повышением дозы, повышать дозировку следует через каждые 5 периодов полувыведения препарата – чтобы уровень содержания препарата в крови стабилизировался перед следующим повышением дозы до 8-10 мг/кг в сутки.
- в комбинации риск побочных реакций у топирамата повышается, влияние на когнитивные функции.
Вальпроат + окскарбазепин:
- синергизм по противосудорожному эффекту;
- назначается при фокальных типах приступов, меньше побочных эффектов чем а карбамазепина и более эфективен;
- фармокинетически окскарбазепин не снижает концентрацию вальпроата в крови;
- начальная доза 8-10 мг/кг в сутки с повышением дозы через каждые 5 периодов полувыведения препарата до 20 мг/кг в сутки.
Вальпроат + этосуксимид:
- синергизм по противосудорожному эффекту;
- используется при лечении генерализованной эпилепсии, абсансной, с феноменом вторичной билатеральной синхронизации;
- исследований по фармакокинетическим взаимодействиям и токсичности нет.
- начальная доза составляет 10 – 25 % от нормальной поддерживающей дозировки.
Повышать дозировку следует через каждые 5 периодов полувыведения препарата – чтобы уровень содержания препарата в крови стабилизировался перед следующим повышением дозы. Если приступы частые, может потребоваться более быстрое повышение дозы.
Карбамазепин + топирамат:
- синергизм по противосудорожному эффекту;
- назначают только при фокальных типах эпилепсии в случае резистентности монотерапией;
- карбамазепин снижает концентрацию топирамата в крови;
- нет данных по нейротоксическим эффектам;
- начальная доза 2 мг/кг в сутки с повышением дозычерез каждые 5 периодов полувыведения до 8-10 мг/кг в сутки.
Карбамазепин / окскарбазепин
- Особенно если приступ начинается с ауры и если возможны комплексные парциальные припадки.
- Окскарбазепин предпочтительнее. Препараты повышают частоту абсансов и миоклонических припадков, поэтому их не следует назначать, если по данным анамнеза можно предположить этот тип припадков.
Все перечисленные выше комбинации представляют собой сочетание так называемого «традиционного» ПЭП (вальпроат, карбамазепин) и одного из «новых» ПЭП (окскарбазепин, ламотриджин, леветирацетам, топирамат). В последнее время стали встречаться комбинации двух «новых» ПЭП. Из экспериментальных исследований известно, что леветирацетам синергичен с окскарбазепином и топираматом, отмечается позитивный нейротоксический профиль. Топирамат с ламотриджином также дают дополнительную эффективность и антагонизм в отношении побочных эффектов, но уже в соотношении доз 1:1. Комбинация окскарбазепина и ламотриджина (так же как комбинация карбамазепина и ламториджина) обладает антагонизмом в отношении противосудорожного эффекта и синергичной нейротоксичностью. Консенсус по оптимальной комбинации ПЭП в лечении резистентной фокальной эпилепсии у детей отсутствует (Белоусова Е.Д., 2014).
Основные противосудорожные препараты.
I. Препараты вальпроевой кислоты (ВПА)
Показания - все формы эпилепсии. Препараты вальпроевой кислоты ‒ антиконвульсанты широкого спектра действия, они эффективны при генерализованных и парциальных приступах, а также при трудно дифференцируемых припадках. Парентеральные формы вальпроатов показаны пациентам любого возраста при лечении всех разновидностей эпилептического статуса.
Противопоказания. Болезни печени (в стадии обострения), наличие в семейном анамнезе случаев тяжелого нарушения функции печени, порфирия, геморрагический диатез.
Взаимодействие. Вальпроаты часто увеличивают концентрацию в плазме крови активных метаболитов карбамазепина и ламотриджина, фенобарбитала, фенитоина (дифенина), иногда увеличивают концентрацию в плазме этосуксимида и примидона.
Побочные эффекты. Тремор, увеличение массы тела, выпадение волос (алопеция), анорексия, диспепсические явления, периферические отеки, сонливость, гипераммониемия (перечисленные побочные эффекты относят к дозозависимым). Возможны острый панкреатит, гепатотоксичность, тромбоцитопения, энцефалопатия.
Предостережения. Вальпроаты с осторожностью назначают при нарушениях функции печени и почек, тенденции к геморрагии, а также перед любыми оперативными вмешательствами.
Дозы и применение при эпилепсии. В педиатрической практике принимают внутрь, начиная с 15‒20 мг/кг, поддерживающая доза ‒ 30‒50 мг/кг (иногда до 80‒100 мг/кг в день). Частота приема ‒ 3 раза в сутки. Пролонгированные («ретард») формы препаратов вальпроевой кислоты рекомендуется 1‒2 раза в сутки. Терапевтическая концентрация в плазме крови у детей ‒ 50‒100 мг/мл (350‒700 мкмоль/л).
Вальпроевая кислота (vа1рrоic асid):
Депакин («Sаnоfi -Sуnthelаbо, Франция): таблетки пролонгированного действия депакин хроно 300 и 500 мг; сироп 57,54 мг/мл во флаконах по 150 мл.
Вальпроат натрия:
*Конвулекс (Gеrоt Рhаrmаreutikа, Австрия): капсулы по 150, 300 и 500 мг; раствор для приема внутрь (капли) 300 мг/мл; сироп для детей 50 мг/мл.
Вальпроат кальция:
Конвульсофин (Arzneimittelwerk Dresden GmbH, Германия; Рlivа, Хорватия) ‒ таблетки по 300 мг.
https://www.cochranelibrary.com/cdsr/doi/10.1002/14651858.CD003032.pub4/epdf/full
II. Габапентин (gаbареptin)
Показания – монотерапия или дополнительная терапия при лечении пациентов старше 12 лет с парциальными припадками с вторичной генерализацией или без таковой. Дополнительная терапия парциальных припадков (в том числе с вторичной генерализацией) у детей от 3 до 12 лет.
Противопоказания. Панкреатит, повышенная чувствительность к препарату. (Габапентин не применяют в виде монотерапии у детей до 12 лет!)
Побочные эффекты. Сонливость, головокружение, слабость, головная боль, атаксия, тремор, диплопия, нистагм, амблиопия, дизартрия, судороги, тошнота, рвота, диспепсия, редко панкреатит, ринит, фарингит, кашель, миалгия, увеличение массы тела, синдром Стивенса ‒ Джонсона.
Предостережения. С острожностью применяют при смешанных формах эпилепсии, включая абсансы, так как под влиянием габапентина возможно усиление их проявления.
Дозы и применение при эпилепсии. Взрослым и детям старше 12 лет: 1-й день ‒ 300 мг/сут, впоследствии суточную дозу ежедневно увеличивают на 300 мг до появления выраженного терапевтического эффекта (максимальная доза ‒ 3,6 г/сут.). Дети в возрасте 3‒12 лет: эффективная доза ‒ 25‒35 мг/кг/сут. в три равных приема. Титровать дозу до эффективной можно в течение трех дней: 10 мг/кг/сут. ‒ в первый день, 20 мг/кг/сут. ‒ второй и 30 мг/кг/сут. ‒ на третий день. Возможно назначение по схеме: при массе тела 17‒25 кг ‒ 600 мг/сут. соответственно при 26‒36 кг ‒ 900 мг/сут., при 37‒ 50 кг ‒ 1200 мг/сут., при 51‒72 кг ‒ 1800 мг/сут. *Нейронтин (Рfi zer, США): капсулы по 0,1; 0,3; 0,4 г; таблетки, покрытые оболочкой, по 0,6; 0,8 г.
https://www.cochranelibrary.com/cdsr/doi/10.1002/14651858.CD010608/epdf/full
III. Карбамазепин (саrbаmаzерinе)
Обладает противоэпилептическим, анальгезирующим, антидепрессивным, а также нормотимическим действием.
Показания - парциальные припадки с вторичной генерализацией (или без таковой), первично-генерализованные тонико-клонические припадки (но не другие генерализованные припадки).
Противопоказания. Нарушения атриовентрикулярной проводимости, нарушения костно-мозгового кроветворения, порфирия, одновременное применение ингибиторов МАО (ацетазоламид) и солей лития, абсансы, миоклонические припадки.
Побочные эффекты. Дозозависимые ‒ диплопия, головокружение, головная боль, тошнота, сонливость, нейтропения, гипонатриемия, гипокальциемия, нарушения ритма сердца. Недозозависимые ‒ кожные высыпания, агранулоцитоз, апластическая анемия, гепатотоксичность, синдром Стивенса ‒ Джонсона, тромбоцитопения.
Предостережения. С осторожностью следует назначать детям при нарушениях функции печени и почек, аллергических реакциях, глаукоме.
Взаимодействие. Несовместим с ингибиторами МАО. Увеличивает гепатотоксичность изониазида. Снижает эффекты антикоагулянтов, противосудорожных средств (производные гидантоина или сукцинамиды), барбитуратов, клоназепама, прими- дона, вальпроевой кислоты. Фенотиазины, пимозид, тиоксантены усиливают угнетение ЦНС (снижается судорожный порог), циметидин, кларитромицин, дилтиазем, верапамил, эритромицин, пропоксифен снижают метаболизм (возрастает риск токсического действия). Понижает активность кортикостероидов, эстрогенов, хинидина, сердечных гликозидов (индукция метаболизма). На фоне ингибиторов карбоангидразы возрастает риск нарушений остеогенеза.
Дозы и применение при эпилепсии. У детей начинать с 8 - 10 мг/кг, поддерживающая доза ‒ 10‒30 мг/кг в день. Обычные формы принимать 2‒3 раза в день, ретардные ‒ 1‒2 раза в день.
Карбамазепин (Ваlkаnрhаrmа, Болгария) ‒ таблетки по 200 мг.
Карбамазепин-Акри («Акрихин», Россия) ‒ таблетки по 200 мг.
Тегретол, тегретол-ЦР (Nоvartis Рhаrmа АG, Швейцария): таблетки по 200 и 400 мг; таблетки-ретард, покрытые оболочкой, по 200 и 400 мг; сироп для орального применения; 2 %-ный раствор (1 мл = 20 мг) во флаконах по 100 мл.
Финлепсин, финлепсин 200 ретард, финлепсин 400 ретард (Arzneimittelwerk Dresden GmbH, Германия; Рlivа, Хорватия) ‒ таблетки по 200 мг, таблетки-ретард по 200 и 400 мг.
https://www.cochranelibrary.com/central/doi/10.1002/central/CN-00070639/full
IV. Клоназепам (сlоnаzераm)
Препарат обладает противоэпилептическим, анксиолитическим, миорелаксирующим, а также снотворным действием.
Показания - Абсансы (хотя клоназепам не является средством выбора вследствие наличия побочных эффектов и вероятного привыкания к препарату). В качестве исходного или дополнительного лечебного средства при атипичных абсансах, атонических, миоклонических припадках.
Противопоказания. Гиперчувствительность к препарату, угнетение дыхательного центра, печеночная и/или почечная недостаточность, миастения, глаукома.
Побочные эффекты. Дозозависимые ‒ утомляемость, седативное действие, сонливость, головокружение, атаксия, возбудимость, агрессивность, гиперкинезы, гиперсаливация, бронхорея, психозы, кожные высыпания, тромбоцитопения.
Предостережения. С особой осторожностью следует назначать детям при нарушении функций печени и почек, дыхания. При резкой отмене препарата возможно усугубление эпилептических припадков.
Взаимодействие. Усиливает влияние нейролептиков, анальгетиков, миорелаксантов. Циметидин пролонгирует эффект. Дифенин увеличивает вероятность возникновения больших судорожных припадков.
Дозы и применение при эпилепсии. Детям в среднем 0,05 ‒0,1 мг/кг/сут. Кратность приема ‒ 3 раза в день.
Клоназепам (Роlfа Польша) ‒ таблетки по 0,5 и 2 мг. Антелепсин (Arzneimittelwerk Dresden GmbH, Германия; Рlivа, Хорватия) ‒ таблетки по 0,25 и 1 мг.
Ривотрил (F.Ноffmаnn-Lа Rосhе Ltd., Швейцария): таблетки по 0,5 и 2 мг; раствор для инъекций (1 мг в 2 мл) в ампулах.
https://www.cochrane.org/ru/CD012253/EPILEPSY_dopolnitelnaya-terapiyaklonazepamom-pri-refrakternoy-epilepsii-u-vzroslyh-i-detey
V. Ламотриджин (lаmоtriginе)
Противоэпилептический препарат, блокирующий потенциалзависимые натриевые каналы, стабилизирующий мембраны нейронов и ингибирующий высвобождение глутаминовой кислоты, играющей ключевую роль в возникновении эпилептических припадков.
Показания-парциальные, комплексные парциальные и генерализованные (первично и вторично) припадки, включая тонико-клонические припадки, а также атонические приступы, абсансы, резистентные к терапии другими антиконвульсантами, припадки, связанные с синдромом Леннокса ‒ Гасто (детям старше 2 лет).
Противопоказания. Гиперчувствительность к препарату, выраженные нарушения функции печени, печеночная недостаточность, возраст до 2 лет (с острожностью назначают детям до 6 лет).
Побочные эффекты. Головная боль, головокружение, сонливость, нарушения сна, ощущение усталости, агрессивность, спутанность сознания, тошнота, дисфункция печени, лимфоаденопатия, лейкопения, тромбоцитопения, кожные высыпания, ангионевротический отек, синдром Стивенса ‒ Джонсона, токсический некроз кожи.
Предостережения. Следует тщательно следить за возможными аллергическими проявлениями, а также контролировать показатели крови и функции печени, не допускать резкой отмены препарата (только при развитии выраженных побочных эффектов). С осторожностью назначать при любых нарушениях печени и почек, аллергических реакциях.
Взаимодействие. Другие противосудорожные средства (карбамазепин, фенитоин, вальпроевая кислота), индуцирующие систему цитохрома P 450 а также парацетамол ускоряют элиминацию.
Дозы и применение при эпилепсии. При терапии ламотриджином и вальпроатом натрия в сочетании с другими антиэпилептическими препаратами или без них детям от 2 до 12 лет: 1‒2-я недели ‒ 0,2 мг/кг/сут., 3‒4-я недели ‒ 0,3 мг/кг/сут. (давать в один прием), для достижения терапевтического эффекта дозу увеличивают на 0,3 мг/кг/сут. через каждые 1‒2 недели до поддерживающей дозы 1‒5 мг/кг, но не более 200 мг/сут. (в 1‒2 приема); при терапии ламотриджином и антиэпилептическими препаратами, инициирующими печеночные ферменты, в сочетании с другими АЭП или без них (за исключением вальпроата натрия): 1‒2-я неделя – 0,6 мг/кг, 3‒4-я недели ‒ 1,2 мг/ кг (в два приема), для достижения терапевтического эффекта дозу увеличивают на 1,2 мг/кг/сут. через каждые 1‒2 недели до поддерживающей дозы 5‒15 мг/кг (в два приема), но не более 400 мг/сут.
Взрослым и детям старше 12 лет: в составе комбинированной терапии при совместном применении Ламиктала и других ПЭП, ингибирующих печеночные ферменты (например, с препаратами вальпроевой кислоты), в течение первых двух недель Ламиктал назначают в дозе 25 мг через день, затем ‒ 25 мг 1 раз в сутки в течение следующих двух недель, на 5-й неделе дозу следует увеличить до 50 мг/сут. в 1‒2 приема. Стабилизирующая доза на 6-й неделе составляет 100 мг/сут. в 1‒2 приема. Доза может быть увеличена до 300 мг/сут. При монотерапии Ламикталом или в составе комбинированной терапии при совместном применении Ламиктала с препаратами лития, бупропионом, оланзапином, окскарбазепином, без применения индукторов или ингибиторов глюкуронизации ламотриджина в течение первых двух недель Ламиктал назначают в дозе 25 мг 1 раз в сутки, на 3‒4-й неделе ‒ 50 мг/сут. в 1‒2 приема, на 5- й неделе ‒ 100 мг/сут. в 1‒2 приема. Стабилизирующая доза на 6-й неделе составляет 200 мг/сут. в 1‒2 приема. После достижения суточной поддерживающей стабилизирующей дозы другие психотропные препараты могут быть отменены.
Ламиктал (GlaxoSmithKline, Великобритания): таблетки по 25, 50, 100 мг; таблетки жевательные/растворимые по 5, 25, 100 мг.
Веро-Ламотриджин («Верофарм», Россия) ‒ таблетки по 25 и 50 мг.
https://www.cochranelibrary.com/cdsr/doi/10.1002/14651858.CD001909.pub3/epdf/full
VI. Окскарбазепин (охсаrbаzерinе)
Показания - парциальные припадки с вторичной генерализацией или без таковой. Первично-генерализованные тонико-клонические припадки. В виде монотерапии или в комбинации с другими АЭП.
Противопоказания. Гиперчувствительность к препарату.
Побочные эффекты. Повышенная утомляемость, астения, головокружение, головная боль, сонливость, амнезия, апатия, атаксия, нарушение концентрации внимания, дезориентация, депрессия, эмоциональная лабильность, нистагм, тремор, тошнота, гипонатриемия, кожные высыпания. Редко ангионевротический отек, аритмия, АВблокада. Переносимость окскарбазепина лучше, чем карбамазепина, но гипонатриемия отмечается чаще.
Предостережения. Повышенная чувствительность к карбамазепину, беременность.
Дозы и применение при эпилепсии. У детей начальная доза ‒ 8‒10 мг/кг/сут. (в два приема), средняя до 20 мг/кг/сут.
Дозу можно увеличивать с интервалами в одну неделю (не более чем в два раза от начальной).
Трилептал (Nоvаrtis Рhаrmа, S.A., Швейцария): таблетки по 150, 300 и 600 мг; суспензия для приема внутрь ‒ 250 мл.
https://www.cochranelibrary.com/cdsr/doi/10.1002/14651858.CD012433.pub2/pdf/CDSR/ CD012433/CD012433.pdf
VII. Топирамат (Tорirаmаtе)
Показания - в составе политерапии при лечении пациентов с парциальными и генерализованными (тонико-клоническими) эпилептическими припадками, включая вторично-генерализованные. Предназначен также для монотерапии указанных эпилептических синдромов.
Противопоказания. Гиперчувствительность к препарату, возраст до 2 лет.
Побочные эффекты. Атаксия, нарушение концентрации внимания, спутанность сознания, головокружение, усталость, парестезии, сонливость и нарушение мышления. Реже встречаются возбуждение, амнезия, анорексия, афазия, диплопия, эмоциональная лабильность, тошнота, нефролитиаз, нистагм, нарушения речи, извращение вкусовых ощущений, нарушения зрения, снижение массы тела.
Предостережения. У пациентов с умеренно или значительно выраженным поражением почек для достижения устойчивых концентраций в плазме может потребоваться 10‒15 дней. У больных с предрасположенностью к нефролитиазу повышается риск образования конкрементов в почках.
Взаимодействие. Фенитоин и карбамазепин снижают содержание топирамата в плазме крови.
Дозы и применение при эпилепсии. При комплексной терапии взрослым и детям старше 12 лет: от 200 до 400 мг/сут. (в два приема), подбор дозы начинают не более чем с 50 мг, принимая препарат на ночь в течение одной недели. В дальнейшем дозу увеличивают на 50 мг (с недельными интервалами), давая 2 раза в день, до достижения клинического эффекта. При монотерапии взрослым и детям: стартовая доза ‒ 0,5‒ 1 мг/кг/сут., увеличение дозы на 0,5‒1 мг/кг/сут. в два приема с интервалом в 1‒2 недели. Максимальная доза у детей до 12 лет может достигать 10 мг/кг/сут.. Мониторирование концентрации топирамата в крови не обязательно.
Топамакс (Jаnssеn СilagFаrmасеutica LDA, Португалия) ‒ таблетки, покрытые оболочкой, по 25, 50, 100, 200, 300 и 400 мг.
https://www.cochranelibrary.com/cdsr/doi/10.1002/14651858.CD001417.pub4/epdf/full
VIII. Фенитоин (рhеnуtоin)
Противоэпилептическое, противосудорожное, антиаритмическое и миорелаксирующее средство.
Показания - генерализованные тонико-клонические, простые и комплексные парциальные, а также психомоторные припадки.
Противопоказания. Гиперчувствительность, нарушение функций печени и почек, сердечная недостаточность, кахексия, порфирия.
Побочные эффекты. Головокружение, возбуждение, головная боль, тремор, атаксия, нистагм, лихорадка, тошнота, рвота, изменения соединительной ткани (огрубение черт лица, контрактуры Дюпюитрена), кожные аллергические реакции (высыпания, зуд), синдром Стивенса ‒ Джонсона, геморрагическая болезнь новорожденных, гепатотоксичность, диспепсия, гиперплазия десен, остеопатии, гипокальциемия, гипергликемия, мегалобластная анемия, лимфоаденопатия, периферическая нейропатия, гирсутизм.
Предостережения. Следует с осторожностью назначать при нарушении функций печени. Необходимо контролировать картину крови (общий клинический и биохимический анализы крови). Не рекомендуется вводить в одной дозе с другими препаратами (во избежание преципитации).
Взаимодействие. Биотрансформацию препарата ускоряют фенобарбитал и карбамазепин, замедляют изониазид и его производные; левомицетин, кумарины, ацетилсалициловая кислота могут усиливать побочные эффекты.
Дозы и применение при эпилепсии. Детям назначают начиная с 5 мг/кг/сут, поддерживающая доза ‒ 6‒8 мг/кг/сут., максимальная доза ‒ 200 мг/сут. Терапевтическая концентрация в плазме крови ‒ 10‒20 мкг/мл.
Дифенин (Россия) ‒ таблетки с содержанием 0,117 г дифенина и 0,032 г натрия гидрокарбоната.
https://www.cochranelibrary.com/cdsr/doi/10.1002/14651858.CD001769.pub3/epdf/full
IX. Препараты группы барбитуратов (фенобарбитал и др.)
Фенобарбитал (рhеnоbаrbitаli)
Обладает противоэпилептическим, седативным, снотворным и миорелаксирующим действием.
Показания. Все виды эпилептических припадков, кроме абсансов.
Противопоказания. Печеночная, почечная (или сочетанная) недостаточность, миастения, гиперчувствительность.
Побочные эффекты. Утомляемость, усталость, ощущение разбитости, астения, бессонница, атаксия, нистагм, гиперкинезы, возбудимость, агрессивность, ухудшение памяти, дефицит фолатов, гипотония, кожная сыпь, эксфолиация, токсический эпидермальный некролиз, гепатоксичность, гипокальциемия, остеомаляция, неонатальные кровотечения, рахит (у детей 1‒2-го года жизни).
Предостережения. Следует назначать препарат с осторожностью при нарушениях функций печени и/или почек, органов дыхания. Следует контролировать картину крови, а также функции печени и почек. (Резкое прекращение приема препарата недопустимо!)
Взаимодействие. Ацетазоламид ослабляет эффект фенобарбитала (тормозит реабсорбцию в почках), седативные средства ‒ усиливают (могут приводить к угнетению дыхания). Фенитоин, производные вальпроевой кислоты повышают содержание фенобарбитала в сыворотке. Фенобарбитал снижает в крови уровень непрямых коагулянтов, кортикостероидов, гризеофульвина, эстрогенов.
Дозы и применение при эпилепсии. Взрослым назначается по 50 мг в день и увеличивается на 25‒50 мг каждый день до эффективной дозы 50‒100 мг 2 раза в день. Детям назначают из расчета 3‒5 мг/кг/сут. (в два приема). Дозы повышают и снижают медленно. Терапевтическая концентрация в плазме крови ‒ 15‒40 мкг/мл. Фенобарбитал (Россия): таблетки по 5, 50 и 100 мг; 0,2 %-ный раствор для приема внутрь (во флаконах по 100 мл).
https://www.cochranelibrary.com/cdsr/doi/10.1002/14651858.CD002217.pub3/epdf/full
XI. Примидон (рirimidоnе)
Противоэпилептический препарат. В печени частично трансформируется в фенобарбитал и фенилэтилмалонамид.
Показания. Эпилепсия различного генеза, большие судорожные припадки; менее эффективен при очаговых, миоклонических, акинетических приступах.
Противопоказания. Гиперчувствительность (в том числе к барбитуратам), печеночно-почечная недостаточнность, анемия, лейкопения.
Побочные эффекты. Сонливость, головокружение, головная боль, апатия, беспокойство, тошнота, рвота, анемия, лейкопения, лимфоцитоз, аллергические реакции.Редко нистагм, атаксия, «волчаночный» синдром, артралгии, психотические реакции, мегалобластная анемия, лекарственная зависимость.
Предостережения. В случае развития мегалобластной анемии следует прекратить прием и начать лечение фолиевой кислотой и/или витамином В12.
Взаимодействие. Усиливает (взаимно) эффект карбамазепина, дифенина и других противосудорожных средств.
Дозы и применение при эпилепсии. Детям назначают по 12‒25 мг/кг/сут. в два приема (поддерживающая доза); начинают с назначения 125 мг (однократно), затем каждые три дня дозу увеличивают на 125 мг (для детей до 9 лет) до достижения необходимого эффекта. Максимальная суточная доза для детей ‒ 1 г (в два приема).
Гексамидин (Россия) ‒ таблетки по 125 и 250 мг.
Это группа барбитуратов
XII. Этосуксимид (еthоsuхimidе)
Противосудорожное, миорелаксирующее, анальгезирующее средство.
Показания. Различные варианты малых припадков первичной и вторичной генерализации, в ряде случаев ‒ при миоклонических приступах.
Противопоказания. Гиперчувствительность, печеночная и почечная недостаточность.
Побочные эффекты. Тошнота, рвота, снижение аппетита, икота, боли в животе, диарея, похудение, головные боли, головокружение, раздражительность, эйфория, фотофобия, кожная сыпь, гирсутизм, нарушение сна, состояние тревоги, невозможность концентрации внимания, агрессивность, редко нарушение кроветворения (нейтропения, агранулоцитоз, эозинофилия, панцитопения, апластическая анемия), альбуминурия, микрогематурия, лекарственная системная красная волчанка, паркинсонизм, миопия. В отдельных случаях параноидально-галлюцинаторные симптомы, развитие больших эпилептических припадков.
Предостережения. Желательно периодическое определение препарата в плазме крови для корректировки дозы (терапевтическая концентрация этосуксимида в крови ‒ 50‒100 мкг/мл).
Взаимодействие. Замедляет биотрансформацию дифенина в печени, повышает его концентрацию в крови (может развиться нистагм, атаксия, гиперплазия десен). Карбамазепин ускоряет метаболизм и снижает содержание этосуксимида в плазме. Значимо изменяет уровень препарата (уменьшает или увеличивает) вальпроевая кислота. Риск развития токсических эпизодов повышает изониазид.
Дозы и применение при эпилепсии. Назначается внутрь, детям ‒ 250 - 500 мг/сут., в 1‒2 приема. Дозу увеличивают постепенно, 1 раз в 4‒7 дней. Максимальная доза ‒ 1,5 г, оптимальная суточная доза для детей ‒ 20 мг/кг.
https://www.cochranelibrary.com/cdsr/doi/10.1002/14651858.CD003032.pub4/epdf/full
XIII. Леветирацетам (кеппра)
Показания. В качестве монотерапии (препарат первого выбора) при лечении парциальных припадков с вторичной генерализацией или без таковойу детей старше 4 лет. С эпилепсией миоклонических судорог, с ювенильной миоклонической эпилепсией, первично-генерализованными (тонико-клоническими) припадками,с идиопатической генерализованной эпилепсией у детейстарше 12 лет.
Противопоказания: нарушение толерантности к фруктозе (для раствора); детский возраст до 4 лет (безопасность и эффективность препарата не установлены); повышенная чувствительность к другим производным пирролидона; повышенная чувствительность к компонентам препарата. С осторожностью следует назначать препарат пациентам с тяжелыми нарушениями функции печени, с почечной недостаточностью, пациентам пожилого возраста (старше 65 лет).
Побочные эффекты. Определение частоты побочных реакций: очень часто (> 1/10), часто (> 1/100, < 1/10), включая отдельные сообщения. Со стороны ЦНС: очень часто сонливость или бессонница, депрессивный синдром, эмоциональная неустойчивость, агрессивность, нервозность; часто головокружение, головная боль, амнезия, атаксия, судороги, гиперкинезия, тремор, диплопия, нарушение аккомодации; в отдельных случаях нарушение поведения, состояние гнева, тревожность, беспокойство, спутанность сознания, галлюцинации, раздражительность, психотические расстройства, самоубийство, попытки самоубийства и мысли о самоубийстве. Со стороны пищеварительной системы: часто абдоминальная боль, диарея, диспепсия, тошнота, рвота, анорексия. Дерматологические реакции: часто кожная сыпь; в отдельных случаях алопеция (во многих случаях восстановление волосяного покрова наблюдалось поcлe отмены препарата).
Предостережения. Пациентам с нарушением функции печени легкой и средней степени тяжести коррекция режима дозирования не требуется. При КК < 70 мл/мин рекомендуется сокращение суточной дозы на 50 %.
Взаимодействие. Возможно совместное применение с другими противоэпилептическими препаратами (фенитоин, карбамазепин, вальпроевая кислота, фенобарбитал, габапентин и примидон).
Дозы и применение при эпилепсии. Принимают внутрь независимо от приема пищи. Суточную дозу препарата делят на два одинаковых приема. В качестве монотерапии детям до года (информированное согласие родителей) начинать с 20 мг/кг/сут., максимальная доза ‒ 35 мг/кг/сут. в два приема. В составе комплексной терапии: детям начинать с 20 мг/кг/сут., максимальная доза 60 мг/кг/сут. в два приема. Изменение дозы на 20 мг/кг массы тела можно осуществлять каждые две недели до достижения рекомендуемой суточной дозы 60 мг/кг (по 30 мг/кг 2 раза в сутки). При непереносимости рекомендуемой суточной дозы возможно ее снижение. Следует применять минимальную эффективную дозу.
Кеппра (UCB Pharma S.A) ‒ таблетки 250, 500, 1000 мг.
https://cyberleninka.ru/article/n/effektivnost-i-perenosimost-levetiratsetama-preparatkeppra-v-lechenii-epilepsii-obzor-literatury/viewer
XIV Вигабатрин (Vigabatrin) / Сабрил (Sabril)
Показания: генерализованные фотосенситивные припадки;
- синдром Веста;
- осложненные судороги фебрильного характера;
- парциальные приступы
Противопоказаниями к применению
- беременность;
- период лактации (грудного вскармливания);
- повышенная чувствительность к какому либо из компонентов препарата;
- у пациентов находящихся в состоянии острого отравления алкоголем, снотворными, обезболивающими, нейролептиками, антидепрессантами, солями лития,
С осторожностью следует назначать препарат пациентам с тяжелыми нарушениями функции печени, с почечной недостаточностью, пациентам пожилого возраста (старше 65 лет).
Взаимодействие: Снижает содержание фенитоина в плазме на 16-33%.
Побочные действия:
Тревожность, раздражительность, депрессия, повышенная агрессивность, психотические расстройства, головная боль, нистагм, тремор, парестезии, снижение способности к концентрации внимания, повышенная утомляемость, дефект полей зрения, атрофия сетчатки, невропатия, атрофия зрительного нерва, диспепсия, увеличение массы тела, отеки. У детей: возбуждение, учащение эпилептических припадков, эпилептический статус; гепатотоксичность, нефротоксичность, кардиотоксичность.
Меры предосторожности
Отменять Сабрил необходимо постепенно, снижая дозу в течение 2–3 нед — резкое прекращение приема может привести к возобновлению судорог. При нарушении полей зрения обязательна консультация офтальмолога. В случае снижения Cl креатинина менее 60 мл/мин следует снизить дозы.
Дозы и применение при эпилепсии:
Внутрь (до или через 2 ч после приема пищи). Взрослым - в суточной дозе 1 г за 1-2 приема. Максимальная суточная доза - 2-4 г. Детям - в суточной дозе 40 мг/кг, максимальная суточная доза - 80-100 мг/кг. При массе тела 10-15 кг - 0.5-1 г/сут, 15-30 кг - 1-1.5 г/сут, 30-50 кг - 1.5-3 г/сут, более 50 кг - 2-4 г/сут. Детям с синдромом Веста - 100 мг/кг. При КК менее 60 мл/мин снижают дозу. При прекращении лечения - постепенная отмена в течение 2-4 нед.
https://www.cochranelibrary.com/cdsr/doi/10.1002/14651858.CD001770.pub3/epdf/full
Таблица 16
АЭП применяемые у пациентов детского возраста [11]
|
Препарат
|
Суточная доза
|
Режим дозирования
|
Поддерживающая доза
|
Терапевтическая концентрация препаратов в плазме мкг/мл
|
|
Карбамазапин
|
Меньше 6 лет: 8-10мг/кг/сут; 6–12 лет: 100 мг (на 2 приема в сут)
|
2 – 3 раза в день
|
10–20 мг/кг/сут
|
4–12
|
|
Клоназепам
|
Меньше 10 лет или масса тела меньше 30 кг: 0,01– 0,2 мг/кг/сут; больше 10 лет: 1– 1,5мг/сут
|
1 – 2 раза в день
|
0,1 – 0,2 мг/кг/ сут
|
0,02 – 0,08
|
|
Ламотриджин
|
В комбинации с ферментиндуцирующими АЭП: - 1мг/кг в сут; в комбинации с вальпроатами 0,3 –0,5мг/кг/сут; в комбинации с ингибиторами ферментов печени 0,3мг/кг/сут; в монотерапии 0,5мг/кг/сут
|
2 – 3 раза в день
|
в комбинации с ферментиндуцирующими 10мг/кг /сут в комбинации с вальпроатами; 5мг/кг/сут 10мг/сут в монотерапии.
|
|
|
Фенобарбитал
|
Меньше года: 3 – 5 мг/кг/сут; больше 1года: 2 – 4 мг/кг/сут
|
1 – 2 раза в день
|
Меньше года: 3 - 5 мг/кг; больше 1года: 2 - 4 мг/кг
|
10 – 40
|
|
Топирамат
|
1 – 3 мг/сут (до 2-х лет возраста по информирова нному согласию родителей)
|
2 раза в день
|
5 – 8 мг/кг/сут
|
-
|
|
Вальпроевая кислота
|
10 – 20мг/кг/сут
|
2 – 3 раза в день
|
30 – 80 мг/кг/сут
|
50 – 100
|
|
Вигабатрин
|
50 мг/кг, максимальная суточная доза - 80-150 мг/кг. При массе тела 10-15 кг – 15-30 кг – 30-50 кг – более 50 кг – Детям с синдромом Веста - 100 мг/кг.
|
0.5-1 г/сут, 1-1.5 г/сут, 1.5-3 г/сут, 2-4 г/сут.
|
|
|
Мониторинг состояния пациента проводится на каждом этапе наблюдения за больным:
• дневник введения приступов;
• видеорегистрация приступа (на телефон);
• контроль общеклинических анализов и ЭЭГ;
• оценка эффективности лечения.
Индикаторы эффективности лечения:
1. Электроклиническая ремиссия: полный контроль – отсутствие приступов в течение более 3х лет и и патологической активности на ЭЭГ;
2. Неполный контроль приступов – редукция частоты приступов более чем на 50%;
3. Неконтролируемые приступы – отсутствие эффекта лечения;
4. Аггравация приступов – аггравации под влиянием ПЭП, предложенная E. Perucca и соавт. в 1998 г., [12], подразделяет этот феномен на два типа:
• тип А – парадоксальная интоксикация – увеличение частоты приступов как проявление чрезмерной для данного пациента лекарственной нагрузки;
• тип В – специфическая (фармакодинамическая) аггравация – специфичный для пациента или типа заболевания феномен, при котором определенные типы приступов появляются, возобновляются или учащаются под влиянием определенных ПЭП в связи с их механизмом действия.
Лечение (скорая помощь)
ЛЕЧЕНИЕ НА ЭТАПЕ СКОРОЙ НЕОТЛОЖНОЙ ПОМОЩИ:
Медикаментозное лечение:
уложить пациента на бок, защита головы от удара во время приступа, расстегнуть ворот, доступ свежего воздуха, подача кислорода. Диазепам 0,5% р-р в/в или в/м (амп 2мл/10мг, ) 0,1 мл/0,5мг/кг; ректально У детей РД не более 2мл. Максимальная суточная доза не более 40мг.
Основной побочный эффект – угнетение дыхания [13].
Вальпроевая кислота р-р в/в амп. 5мл №5 (100мг/мл) В/в медленно РД 15мг/кг в течении 5 мин. 20мг/кг/сут у взрослых.
Лечение (стационар)
ДИАГНОСТИКА И ЛЕЧЕНИЕ НА СТАЦИОНАРНОМ УРОВНЕ:
Диагностические критерий на стационарном уровне:
Жалобы и анамнез:
Физикальное обследование:
Лабораторные исследования:
• общий анализ крови – при длительном приеме антиэпилептических препаратов, могут возникнуть побочные действия со стороны крови: снижение количества тромбоцитов, снижение свертываемости крови;
• биохимический анализ крови (глюкозы, АЛТ, АСТ, тимоловая проба, билирубин общий, амилаза, щелочная фосфатаза, общий белок, мочевина, креатинин (по показаниям).
Инструментальные исследования:
• Электроэнцефалография – рутинная, позволяет определить наличие патологической электрической активности, указывает на расположение эпилептического очага;
• Длительное ЭЭГ - мониторирование – для уточнения типа приступа и регистрации иктального ЭЭГ;
• МРТ головного мозга – для выявления органической патологии;
• КТ головного мозга – при отсутствии МРТ;
Перечень дополнительных диагностических мероприятий:
• Нейросонография;
• Видеомониторинг электроэнцефалограммы (1-3-5 часов);
• Ночной видеомониторинг электроэнцефалограммы;
• УЗИ органов брюшной полости;
• МРТ головного мозга (по показаниям) ПО эпилептологическому протоколу.
Метод диагностики входящий в структуру предоперационного обследования, фармокорезистентных форма эпилепсии, для диагностики мезиального темпорального склероза, сосудистых мальформаций, дисгенезий мозга, дифференцировка патологического участка между ФКД и глиальными образованиями, при метаболических, митохондриальных энцефалопатиях и при очагах неясной этиологии;
• Исследование спинномозговой жидкости (по показаниям): при подозрении на инфекцию или опухоль мозга;
• Определение лактатдегидрогиназы (ЛДГ);
• Определение кальция (Ca) в сыворотке крови;
• Определение лактата (молочной кислоты) в сыворотке крови;
• определение уровня АЭП в крови;
• определение уровня альфафетопротеина (АФП).
Мониторинг состояния пациента:
• дневник введения приступов;
• видеорегистрация приступа (на телефон);
• контроль общеклинических анализов и ЭЭГ.
Дальнейшее ведение на амбулаторном уровне (ЦСМ):
• пациенту необходимо заполнять дневник наблюдений за симптомами и показывать его при каждом визите врачу.
• для предупреждения побочных эффектов необходим контроль за показателями общего и биохимического анализа крови 1 раз в 3 или 6 месяцев в зависимости от клиники и типа препарата.
• родственникам больного необходимо помнить о возможности развития эпилептического приступа и, по возможности, стремиться обеспечить условия, при которых приступ (при возникновении) был бы наименее травматичным, а также уметь оказать первую неотложную помощь.
• При благоприятном течении заболевания рекомендуется посещение врача 2 раза в год с результатами ЭЭГ от 2 до 5-ти лет от момента последнего приступа в зависимости от формы эпилепсии с последующим снятием с учета. При неблагоприятном течении заболевания посещение врача определяется частотой приступов.
• Проведение медико-социальной экспертизы
Госпитализация
ПОКАЗАНИЯ ДЛЯ ГОСПИТАЛИЗАЦИИ СУКАЗАНИЕМ ТИПА ГОСПИТАЛИЗАЦИИ:
1 Показания для плановой госпитализации:
• впервые возникшие эпилептические приступы у детей (для выявления этиологических факторов, уточнения характера, частоты приступов, подбора препаратов, отработки адекватной схемы лечения);
• при неэффективности амбулаторного лечения;
• появление у больного других неврологических симптомов; увеличение частоты приступов или изменение их стереотипа;
2 Показания для экстренной госпитализации:
• учащение приступов;
• эпилептический статус, серийные приступы, сумеречное расстройство сознания;
• необходимость хирургического лечения (в нейрохирургический стационар).
Информация
Источники и литература
-
Клинические протоколы Министерства здравоохранения Кыргызской Республики
- Клинические протоколы Министерства здравоохранения Кыргызской Республики - 1. ICD coding for epilepsy: Past, present, and future. A report by the International League Against Epilepsy Task Force on ICD codes in epilepsy. Nathalie Jette, Ettore Beghi et all. Epilepsia, 56(3):348–355, 2015; 2. Robert S.Fisher et al Epileptic seizure and Epilepsy: Definitions Proposed by ILAE and IBE. Epilepsia 46(4); 470-472, 2005; 3. Definition of drug resistant epilepsy: Consensus proposal by the ad hoc Task Force of the ILAE Commission on Therapeutic Strategies. Patrick Kwan, Alexis Arzimanoglou et all. Epilepsia, 51(6):1069–1077, 2010; 4. A practical clinical definition of epilepsy. Robert S. Fisher et al. Epilepsia, 55(4):475–482, 2014; 5. Commission classification and terminilogy of the International League Against Epilepsy. Proposal for revised clinical and electroencephalographic classification of epileptic seizures. Epilepsia, 1981;22:489-501; 6. Revised terminology and concepts for organization of seizures and epilepsies: Report of the ILAE Commission on Classification and Terminology, 2005–2009. Anne T. Berg, Samuel F. Berkovic, Martin J. Brodie, J. Helen Cross et all. Epilepsia, 51(4):676–685, 2010; 7. The Epilepsies. The diagnosis and management of the epilepsies in adults and children in primary and secondary care. National Clinical Guidelines Center and National Institute clinical Excelllence. 2012; 8. Clinical Aspects of the Ketogenic Diet. Adam L. Hartman and Eileen P. G. Vining. Epilepsia, 48(1):31–42, 2007; 9. Updated ILAE evidence review of antiepileptic drug efficacy and effectiveness as initial monotherapy for epileptic seizures and syndromes. Tracy Glauser et all for the ILAE subcommission of AED Guidelines. Epilepsia, 1–13, 2013; 10. Diagnosis and management of epilepsy in adults. SIGN (Scottish Intercollegiate Guidelines Network) 2015; 11. The Epilepsies. The diagnosis and management of the epilepsies in adults and children in primary and secondary care. National Clinical Guidelines Center and National Institute clinical Excelllence. 2012; 12. Antiepileptic drugs as a cause of worsening seizures. Perucca E1, Avanzini G, Dulac O. Epilepsia. 1998 Jan;39(1): 5-17; 13. Мухин К.Ю., Петрухин А.С. Идиопатические формы эпилепсии. М., 2000, с. 3-12; 14. Presurgical evaluation and outcome of epilepsy surgery in childhood. J. HELEN CROSS.UCL Institute of Child Health, Great Ormond Street Hospital for Children NHS Trust, 2012; 15. Engel J. Approaches to localization of the epileptogenic lesion. In : Engel J Jr ed. Surgical treatment of the epilepsies. New York: Raven Press 1987; 75-95. 16. Hiroki Nariai, MD1, Susan Duberstein, MD1,2, and Shlomo Shinnar, MD, PhD1,2,3. Treatment of Epileptic Encephalopathies: Current State of the Art. J Child Neurol. 2018 January ; 33(1): 41–54. 17. Kenou van Rijckevorsel Reference Centre of Refractory Epilepsy, Cliniques Universitaires St Luc, Université Catholique de Louvain, Brussels, Belgium. Treatment of Lennox-Gastaut syndrome: overview and recent findings. Neuropsychiatric Disease and Treatment 2008:4(6) 1001–1019 18. Sharika V. Raga, Jo M. Wilmshurst. Epileptic spasms: Evidence for oral corticosteroids and implications for low and middle income countries. 1059-1311. 2018 British Epilepsy Association. Published by Elsevier Ltd. All rights reserved.
Информация
Адрес для переписки с рабочей группой
720020, Кыргызская Республика,
г. Бишкек Ул. Ахунбаева 190,
НЦОМиД. Кафедра факультетской педиатрии им И.К Ахунбаева.
bnurmuhamed@gmail.com
СПИСОК ИСПОЛЬЗОВАННЫХ СОКРАЩЕНИЙ
ILAE – International League against Epilepsy
NICE – National Institute for Health and Clinical Excellence
LTME – Long-term monitoring EEG
ППП – простой парциальный приступ
СПП – сложный парциальный приступ
ФЭ – фокальная эпилепсия
ГЭ – генерализованная эпилепсия
ФРЭ – фармакорезистентная эпилепсия
ГСЭП – генерализованный судорожный приступ
ВГСЭП – Вторично–генерализованный судорожный приступ
ЖКТ – желудочно-кишечный тракт
ОАК – общий анализ мочи
ОАМ – общий анализ крови
УЗИ – ультразвуковое исследование
EL (Evidence level) – уровень доказательности
ПЭТ – Позитронно–эмиссионная томография
ОФЭТ – Однофотонно–эмиссионная томография
ЭС - эпилептический статус
УД - уровень доказательности
ГСВ - группы семейных врачей
ЦСМ – центр семейных врачей
Термины, использованные в клиническом руководстве и их определения
Качество жизни - интегральная характеристика физического, психологического, эмоционального и социального функционирования личности, основанная на ее субъективном восприятии.
Копинг-поведение - набор повторяющихся поведенческих стратегий, направленный на преодоление проблем и жизненных трудностей, на преодоление внутреннего напряжения и беспокойства. Вырабатывается на основании собственного жизненного опыта, а также благодаря психосоциальным вмешательствам.
Ребенок – универсальный термин, объединяющий понятия «малолетний», «несовершеннолетний», «дошкольник», «подросток». Ребенком, в соответствии с международным правом, считается лицо от рождения до 18 лет
Психологическое консультирование в контексте данного руководства – профессиональная помощь ребенку и его/ее семье с целью снижения уровня дистресса и восстановления достойного уровня качества жизни после адаптации к диагнозу.
Психолог - лицо, имеющее университетское образование по специальности «психология», а также лица, которым в установленном порядке присвоены ученые степени кандидата наук по специальностям психологической науки.
Прикладной анализ поведения изучает влияние окружающей обстановки на поведение. Под «поведением» понимаются любые действия, совершаемые субъектом. Возможность практического использования законов поведения, но и существование развитой системы помощи детям с эпилепсией и эпилептическими синдромами, и другими пароксизмальными состояниями.
Правовую основу данного клинического руководства составляют:
-
Конституция Кыргызской Республики;
-
Закон Кыргызской Республики «Об охране здоровья граждан»;
-
Закон Кыргызской Республики «О психиатрической помощи и гарантиях прав граждан при ее оказании»;
-
Кодекс Кыргызской Республики о детях;
-
Конвенция о правах ребенка;
-
Семейный кодекс Кыргызской Республики;
Состав рабочей группы
Исполнители:
|
Бабаджанов Н.Д.
|
к.м.н., невролог НЦОМиД.
|
|
Маткеева А.Т.
|
Зав. отделением неврологии НЦОМиД.
|
|
Василенко В.В..
|
К.м.н. Доцент кафедры неврологии, нейрохирургии и медицинской генетики КРСУ им. Б.Н Ельцина.
|
|
Жихарева В.В
|
Зав. отделением ОППП НЦОМиД
|
|
Эркинбек у Н
|
Врач невролог отделения неврологии
|
Консультанты
|
Кадырова Т.М.
|
К.м.н. Доцент кафедры психиатрии, наркологии и клинической психологии КГМА им. И.К. Ахунбаева
|
|
Хамзина А. Р.
|
Детский невролог-эпилептолог.
|
|
Галако Т.И.
|
К.м.н. Доцент Зав. кафедрой психиатрии, наркологии и клинической психологии КГМА им. И.К. Ахунбаева
|
Внутренние рецензенты:
|
Нурбекова У.А.
|
К.м.н. Доцент кафедры неврологии и клинической генетики КГМА им. И.К.Ахунбаева.
|
|
|
|
Внешний рецензент
|
Жусупова А.Т
|
К.м.н. И.о доцента кафедры неврологии и клинической генетики КГМА им. И.К. Ахунбаева.
|
Все вышеперечисленные специалисты работали и работают c детьми и подростками, в практике которых встречались случаи лечения и диагностирования больных эпилепсией и эпилептическими синдромами. Приглашение консультантов в состав разработчиков позволило обсудить вопросы применимости руководства в организациях всех уровней здравоохранения Кыргызской Республики. Протоколы согласительных заседаний мультидисциплинарной рабочей группы по разработке клинического руководства велись на базе НЦОМиД и кафедры факультетской педиатрии КГМА им.И.К. Ахунбаева. Все члены группы подписали декларацию о конфликте интересов.
Процесс утверждения клинического руководства
В 2020 г. клиническое руководство было апробировано в пилотных организациях КР - в центрах семейной медицины Бишкека, Ошской и Джалал-Абадской областей. В процессе апробации клинического руководства были получены комментарии и замечания по форме изложения руководства, которые были учтены при его доработке.
Были получены рецензии от Нурбековой У.А., к.м.н., доцента кафедры неврологии и клинической генетики КГМА им. И.К. Ахунбаева.
Жусуповой А.Т., к.м.н., и.о доцента кафедры неврологии и клинической генетики КГМА им. И.К. Ахунбаева.
После апробирования данное клиническое руководство было утверждено Экспертным советом по оценке качества клинических руководств Министерства здравоохранения Кыргызской Республики.
Методологическая экспертная поддержка
|
Джакубекова А.У
|
Главный специалист по лекарственной политике УОМПиЛП МЗ КР, к.м.н., доцент
|
|
Зурдинова А.
|
внештатный клинический фармаколог МЗ КР, зав. кафедрой базисной и клинической фармакологии КРСУ, доцент, к.м.н., специалист по доказательной медицине
|
Экспертами проводилась методологическая поддержка при разработке клинического руководства и экспертная оценка методологии создания клинического руководства, основанного на принципах доказательной медицины.
ОПИСАНИЕ ПРОЦЕССА ПОИСКА ОЦЕНКИ ДОКАЗАТЕЛЬСТВ И ФОРМУЛИРОВАНИЯ РЕКОМЕНДАЦИЙ
Все основные рекомендации в данном руководстве имеют свою градацию, которая обозначается латинской буквой от A до D (SIGN). При этом каждой градации соответствует определённый уровень доказательности данных, и это значит, что рекомендации основывались на данных исследований, которые имеют различную степень достоверности. Чем выше градация рекомендации, тем выше достоверность исследований, на которой она основана. Ниже приведена шкала, которая описывает различные уровни градации рекомендаций, включённых в данное руководство.
Уровни доказательности по SIGN-50:
Таблица №1
|
A |
- Высококачественный мета-анализ, систематический обзор РКИ или крупное РКИ с очень низкой вероятностью (++) систематической ошибки, результаты которых могут быть распространены на соответствующую популяцию.
|
|
B |
- Высококачественный (++) систематический обзор когортных или исследований случай-контроль или
- Высококачественное (++) когортное или исследование случай- контроль с очень низким риском систематической ошибки или - РКИ с невысоким (+) риском систематической ошибки, результаты которых могут быть распространены на соответствующую популяцию. |
|
C |
- Когортное или исследование случай-контроль или контролируемое исследование без рандомизации с невысоким риском систематической ошибки (+), результаты которых могут быть распространены на соответствующую популяцию или
- РКИ с очень низким или невысоким риском систематической ошибки (++ или +), результаты которых не могут быть непосредственно распространены на соответствующую популяцию |
|
D |
- Описания серии случаев ИЛИ
- Неконтролируемое исследование ИЛИ - Мнение экспертов - Рекомендации, основанные на клиническом опыте членов мульти- дисциплинарной группы |
Поиск клинических руководств осуществлялся в национальных и международных электронных базах данных в сети Интернет.
Крупнейшие электронные базы данных по клиническим руководствам и практическим рекомендациям
Таблица№2
|
Страна и название ресурса
|
Интернет-адрес
|
|
Международная ассоциация
|
|
|
Guidelines International Network (G-I-N)
|
|
|
Доказательство по ментальному здоровью (Evidence of Evidence-based Mental Health)
|
|
|
Society of Clinical Psychiatrists
|
|
|
Соединенные Штаты Америки
|
|
|
US National Guideline Clearinghouse (NGC)
|
|
|
Американская психиатрическая ассоциация
|
|
|
Канада
|
|
|
Canadian Psychiatric Association (CPA)
|
|
|
Великобритания
|
|
|
National Institute for Clinical Excellence (NICE)
|
|
|
Clinical Knowledge Summaries (CKS)
|
|
|
Scottish Intercollegiate Guidelines Network (SIGN)
|
|
|
eGuidelines
|
|
|
National electronic Library for Health
|
|
|
Новая Зеландия
|
|
|
New Zealand Guidelines Group (NZGG)
|
|
|
Россия
|
|
|
Межрегиональное общество специалистов доказательной медицины (ОСДМ)
|
|
|
Клинические рекомендации РФ
|
|
Были установлены языковые ограничения, так как рабочая группа имела возможность изучать источники литературы только на английском и русском языках.
РЕКОМЕНДАЦИИ РОДИТЕЛЯМ ДЕТЕЙ, СТРАДАЮЩИХ ЭПИЛЕПСИЕЙ
Родители, у которых есть дети, страдающие эпилепсией, должны знать особенности этой болезни в целом и приступов, в частности; уметь самим и научить своих близких и родных оказывать первую помощь ребенку при эпилептическом припадке. Необходимо оставаться спокойным и не паниковать. Не давать нашатырный спирт или другие лекарства. Передвигают пациента только для того, чтобы изолировать его от любых повреждающих предметов. Зафиксировать продолжительность судорог по часам с проведением видеорегистрации (визуализация приступов). Повернуть голову или всего ребенка на бок во избежание аспирации слюны или западения языка. Необходимо находиться рядом до окончания приступа.
При приступах эпилепсии могут быть:
Судороги. Во время тонических судорог следует аккуратно, но достаточно твердо придерживать ручки и ножки, чтобы ребенок случайно не навредил сам себе – не поцарапался или не стукнулся обо что-нибудь.
Родителям крайне желательно несколько минут не беспокоить своего ребенка, а дать ему проснуться самостоятельно. После окончания приступа соблюдать следующие правила:
Проверить дыхание - сразу же после исчезновения судорог срочно проверить, дышит ли ребенок. В том случае, если ребенок не дышит, необходимо немедленно начинать проведение искусственного дыхания. Недопустимо начинать реанимационные мероприятия непосредственно во время эпилептического припадка.
Быть рядом. Оставаться рядом до тех пор, пока ребенок полностью не придет в себя.
Не давать жидкость и лекарства до тех пор, пока ребенок полностью не оправится от эпилептического приступа. В противном случае существует риск асфиксии. Следить за температурой. В том случае, если после эпилептического приступа у ребенка поднимается температура, поставить ему ректальную свечку, содержащую парацетамол.
При серийном течении приступов, между которыми ребенок не приходит в сознание, затяжном припадке (более 15 минут) с нарушением дыхания, сердечной деятельности самостоятельно лучше не действовать, а как можно быстрее вызвать бригаду «скорой помощи».
Родители обязаны помочь больному ребенку адаптироваться к жизни в семье, в коллективе, в общении с другими детьми и взрослыми.
Очень важно уделить достаточное количество времени морально-психологической стороне проблемы эпилепсии. Детские психологи считают, что ребенок должен иметь представление о своей болезни и о том, что с ним происходит. Разумеется, информация должна быть подана ребенку в доступной для его понимания форме. Необходимо позаботиться и о том, чтобы коллектив, в котором находится больной ребенок, был в курсе, что малышу может в любой момент стать плохо, и он потеряет сознание. Не стоит скрывать данную информацию от окружающих, так как всегда существует риск того, что приступ эпилепсии случится у ребенка вне дома. Да и взрослые люди, окружающие ребенка – преподаватели в школе, воспитатели в детском саду, смогут оказать необходимую ребенку первую помощь только в том случае, если они обладают необходимой информацией и знают о таком явлении, как эпилепсия у детей.
Обязательно следить за тем, какой образ жизни ведет ваш ребенок и как соблюдает режим дня.
Добиться того, чтобы:
- ребенок ложился спать в определенное время и имел 8-10 часовой ночной сон;
- ограничить время просмотра телевизора, а в некоторых случаях – исключить данную нагрузку, помнить, что игры на компьютере, светомузыка могут провоцировать эпилептические припадки;
- избегать значительных физических и психических перегрузок; включая опасные для жизни занятия спортом (плавание, вело- и мотоспорт), вождение автомобиля, работа на высоте и т.д.);
- исключить употребление алкоголя и курение.
И самая главная задача, которая стоит перед всеми родителями детей, страдающих от эпилепсии – убедить своего ребенка в том, что в его болезни нет ничего страшного или постыдного. И как только он поймет это, то исчезнет огромное количество психологических проблем.
Необходимо всегда помнить, что эпилепсия – не смертный приговор. Адаптация детей с этой болезнью к жизни в детском возрасте – залог их успешной и вполне комфортной взрослой жизни.
Прикреплённые файлы
Внимание!
- Занимаясь самолечением, вы можете нанести непоправимый вред своему здоровью.
- Информация, размещенная на сайте MedElement и в мобильных приложениях "MedElement (МедЭлемент)", "Lekar Pro", "Dariger Pro", "Заболевания: справочник терапевта", не может и не должна заменять очную консультацию врача. Обязательно обращайтесь в медицинские учреждения при наличии каких-либо заболеваний или беспокоящих вас симптомов.
- Выбор лекарственных средств и их дозировки, должен быть оговорен со специалистом. Только врач может назначить нужное лекарство и его дозировку с учетом заболевания и состояния организма больного.
- Сайт MedElement и мобильные приложения "MedElement (МедЭлемент)", "Lekar Pro", "Dariger Pro", "Заболевания: справочник терапевта" являются исключительно информационно-справочными ресурсами. Информация, размещенная на данном сайте, не должна использоваться для самовольного изменения предписаний врача.
- Редакция MedElement не несет ответственности за какой-либо ущерб здоровью или материальный ущерб, возникший в результате использования данного сайта.