Нарушения митохондриального β-окисления жирных кислот
![]() Версия: Клинические рекомендации РФ 2024 (Россия)
Версия: Клинические рекомендации РФ 2024 (Россия)
Общая информация
Краткое описание
- Ассоциация медицинских генетиков
- Союз педиатров России
- Общероссийская общественная организация содействия развитию неонатологии «Российское общество неонатологов»
Одобрено Научно-практическим Советом Минздрава РФ
– размещенные в Рубрикаторе после 1 января 2024 года – с 1 января 2025 года.
Клинические рекомендации
Возрастная категория: Взрослые, Дети
Пересмотр не позднее: 2026
ID: 694
Наследственные нарушения митохондриального β-окисления жирных кислот (FAOD)– группа моногенных заболеваний, связанных с нарушением митохондриального β-окисления и транспорта карнитина и жирных кислот в митохондриях.
Классификация
Классификация заболевания или состояния (группы заболеваний или состояний)
По ведущему симптомокомплексу, выделяют следующие формы FAOD:
По срокам появления первых признаков различают формы FAOD:
Этиология и патогенез
Этиология и патогенез заболевания или состояния (группы заболеваний или состояний)
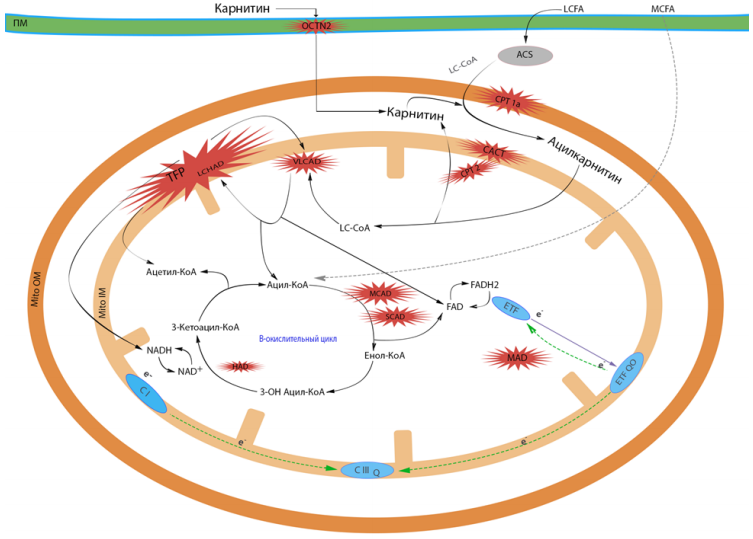
TFP – митохондриальный трифункциональный белок; ПM – плазматическая мембрана; СI,CIII – комплексы дыхательной цепи митохондрий; MitoOM – внешняя митохондриальная мембрана; MitoIM – внутренняя митохондриальная мембрана; ETF – электронпереносящий флавопротеин; CPT1 - карнитин пальмитоилтрансфераза 1; CAСT – карнитин ацилкарнитин транслоказа; CPT2 – карнитин пальмитоилтрансфераза 2; LCFA– длинноцепочечные жирные кислоты; MCFA – среднецепочечные жирные кислоты; OCTN2 – транспортер карнитина плазматической мембраны; ETF QO – ETF(электронпереносящий флавопротеин )-дегидрогеназа.
Эпидемиология
Эпидемиология заболевания или состояния (группы заболеваний или состояний)
Клиническая картина
Cимптомы, течение
Клиническая картина заболевания или состояния (группы заболеваний или состояний)
FAOD имеют много общих клинических проявлений. Обычно выделяют неонатальную (раннюю), детскую (инфантильную) и позднюю (взрослую) формы болезни. Ранняя форма отличается особенной тяжестью, метаболические нарушения часто приводят к нарушению сознания, хотя коматозные состояния могут возникнуть и при детской, и при поздней формах.
Частыми симптомами FAOD являются некетотическая гипогликемия, кардиомиопатия и миопатия, эти симптомы могут проявляться как изолированно, так и в сочетании.
Нарушения сердечного ритма описаны практически при всех FAOD и встречаются в сочетании с кардиомиопатией или изолированно [11].
Миопатия очень частый симптом при нарушениях FAOD. Пациенты могут предъявлять жалобы на миалгию, быструю утомляемость, непереносимость физической нагрузки. Чаще всего миопатия манифестирует в подростковом возрасте, на 2-3 десятилетии жизни, но может быть ведущим симптомом и ранее. Иногда миопатия может провоцироваться интенсивными физическими упражнениями, но также может наблюдаться после анестезии и вирусной инфекции [1-5].
Нарушения транспорта и метаболизма карнитина и жирных кислот
В группу входит несколько заболеваний, различающихся по срокам манифестации, тяжести течения и биохимическим характеристикам.
Транспорт длинноцепочечных ацил-КоА в митохондрии осуществляется с помощью карнитинового цикла, включающего 4 белка: 1) карнитиновый транспортер плазматической мембраны (OCTN2), обеспечивающий внутриклеточную доставку карнитина; 2) карнитин-пальмитоилтрансферазу 1 (СРТ 1) на внешней мембране митохондрий, которая переносит ацильные остатки ЖК от КоА к карнитину; 3) карнитин/ ацилкарнитин транслоказу (САСТ), обеспечивающую челночный транспорт ацилкарнитинов через внутреннюю мембрану в обмен на свободный карнитин; 4) карнитин-пальмитоилтрансферазу 2 (СРТ 2), которая переносит на внутренней мембране митохондрий ацильные остатки ЖК от карнитина к КоА. Мутации соответствующих генов, кодирующих данные белки, приводят к наследственным нарушениям транспорта карнитина и жирных кислот.
Начало заболевания — обычно на первом году жизни, или в младенчестве. Обычно у пациентов отмечается незначительное увеличение печени, и заболевание протекает в виде острых метаболических приступов, как Рейе-подобный синдром, сопровождающихся тахипноэ, рвотой, отказом от еды, судорогами, летаргией, комой. Лабораторно выявляют повышение уровня трансаминаз, удлинением протромбинового и тромбопластинового времени.
Нередко заболевание протекает под маской сепсиса
Аутосомно-рецессивное состояние, связанное с мутациями гена ACADS, кодирующего ацил-КоА дегидрогеназу жирных кислот с короткой углеродной цепью (SCAD).
Аутосомно-рецессивное заболевание, связанное с мутациями гена HADH, кодирующем короткоцепочечную 3-гидроксиацил- КоА дегидрогеназу жирных кислот (SCHAD).
Позднее установление диагноза может привести к формированию эпилепсии и умственной отсталости из-за повторяющейся гипогликемии [77].
Аутосомно-рецессивное заболевание, связанное с мутациями гена ACADM, кодирующего ацил-CoA-дегидрогеназу среднецепочечных жирных кислот (MCAD).
Заболевание в большинстве случаев манифестирует в возрасте от 3-х до 24-х месяцев. Нередко (от 10 до 40% случаев) заболевание начинается в периоде новорожденности. Приступы протекают тяжело и могут заканчиваться летально. В 5% случаев смерть детей происходит в первые дни жизни. Около 20% пациентов умирают до установления диагноза [11, 17]. В большинстве случаев основными проявлениями являются гипокетотическая гипогликемия, которая может сочетаться с лактат-ацидозом и гипераммониемией. Клинические проявления включают синдром угнетения ЦНС, сонливость и рвоту на фоне инфекционных заболеваний, голодания или при оперативных вмешательствах. У некоторых пациентов отмечается метаболический криз, несмотря на избыточное накопление кетоновых тел и нормальную концентрацию глюкозы в крови. Изредка пациенты демонстрируют во время приступа “парадоксально” выраженный кетоз. В период криза отмечается сонливость, рвота, дыхательная недостаточность, судороги, гепатомегалия, а также быстрое развитие сердечной недостаточности. Повреждение головного мозга, которое может возникнуть во время метаболического криза, увеличивает риск отдаленных неврологических нарушений. Может наблюдаться специфическая полиморфная желудочковая тахикардия в виде характерных изменений на ЭКГ в виде torsades de pointes (фр. — «скрученные шнурки»), или желудочковая тахикардия типа «пируэт». Направление комплексов QRS меняется циклически: несколько комплексов направлено вниз, затем в том же отведении —вверх. При объективном обследовании определяется увеличение печени, синдром цитолиза (Рейе-подобный синдром). Межприступный период протекает благоприятно, у 80% пациентов отмечается нормальное нервно-психическое развитие с полным отсутствием других признаков патологии [15, 17, 48, 49].
Аутосомно-рецессивное заболевание, связанное с мутациями гена ACADVL, кодирующего ацил-КоА дегидрогеназу жирных кислот с очень длинной углеродной цепью (VLCAD).
Дефицит митохондриального трифункционального белка (TFP)/недостаточность длинноцепочечной З-ОН ацил КоА дегидрогеназы жирных кислот (LCHAD) / недостаточность длинноцепочечной 3-кето-КоА тиолазы (LCKAT).
Глутаровая ацидурия 2-го типа (множественный дефицит ацил-КоА дегидрогеназ)
Дефицит переносчика рибофлавина
Для терапии синдрома Брауна-Виалетто-Ван Лаэра и болезни Фацио-Лонда высокоэффективен рибофлавин (биологическая активная добавка) в высоких дозах, что требует проведения исследований для дифференциальной диагностики данных нарушений с ГА2 [125].
Дефицит FAD-синтазы (FADS) был впервые описан в 2016 году в 7 неродственных семьях, с неврологическими проявлениями и биохимическим профилем, указывающим на ГА2 [126]. При лечения этой патологии также применяют рибофлавин (биологическая активная добавка).
Диагностика
Диагностика заболевания или состояния (группы заболеваний или состояний) медицинские показания и противопоказания к применению методов диагностики
1. Жалобы и анамнез
- Отягощенный семейный анамнез (сходные симптомы у родных братьев и сестер пробанда, близкородственный брак);
- Случаи внезапной детской смерти;
- HELLP — синдром или жировая дистрофия печени у женщины во время беременности;
- Рвота (в любом возрасте);
- Судороги (в любом возрасте);
- Изменение цвета мочи (красновато-бурая моча) (в любом возрасте);
- Эпизоды нарушения сознания (в любом возрасте);
- Эпизоды нарушений сердечного ритма (в любом возрасте);
- Непереносимость длительных голодных промежутков;
- Непереносимость физической нагрузки (взрослые и дети старшего возраста);
- Мышечные боли (взрослые и дети старшего возраста);
- Быстрая утомляемость (взрослые и дети старшего возраста);
- Эпизоды гипокетотической гипогликемии в анамнезе.
2. Физикальное обследование
- Сонливость, вялость;
- Эпизоды нарушения сознания;
- Судороги;
- Мышечная гипотония;
- Мышечная слабость;
- Боли в мышцах при пальпации;
- Увеличение размеров печени;
- Нарушение сердечного ритма;
- Признаки сердечной недостаточности.
3. Лабораторные диагностические исследования
- Рекомендовано исследование уровня свободного карнитина и ацилкарнитинов в крови всем пациентам с клиническими симптомами, характерными для FAOD [1,105,144].
- Рекомендовано всем пациентам с клиническими симптомами характерными для FAOD определение содержания органических кислот в моче методом газовой хроматографии масс-спектрометрии для исключения других заболеваний из класса наследственных болезней обмена веществ и диагностики ГА2 [6,13].
- Рекомендовано всем пациентам при выявлении биохимических изменений, свидетельствующих в пользу FAOD определение вариантов генов в образце биологического материала другом или неуточненном, неклассифицированные в других рубриках, методом секвенирования по Сенгеру (03.Я99.18.999.039) и/или методом таргетного высокопроизводительного секвенирования (03.Я99.18.998.041) и/или методом высокопроизводительного секвенирования (03.Я99.18.999.041) и/или методом полимеразной цепной реакции (03.Я99.18.999.048) и/или методом множественной лигазно-зависимой амплификации зондов (03.Я99.18.999.059) (выявление биаллельных патогенных вариантов в генах ACADM, ACADVL, HADH, ACADS, HADHA, HADHB, ETFA, ETFB, ETFDH, SLC52A3, SLC52A2, CPT2, CPT1A, SLC25A20, SLC22A5, SLC25A32, ACAD9, ECHS1 и др.) для подтверждения диагноза и проведения медико-генетического консультирования семьи [9].
- Рекомендовано всем пациентам с клиническими и/или биохимическими признаками FAOD общий (клинический) анализ крови развернутый (гемоглобин, количество эритроцитов, цветовой показатель, количество лейкоцитов, тромбоцитов, лейкоцитарная формула и скорость оседания эритроцитов) для оценки основных параметров кроветворения и наличия воспалительных процессов [1, 2, 5].
- Рекомендовано всем пациентам с клиническими и/или биохимическими признаками FAOD проведение анализа крови биохимического общетерапевтического (содержание глюкозы, общего белка, белковых фракций, альбумин, С-реактивного белка, исследование уровня общего билирубина в крови, Исследование уровня свободного и связанного билирубина в крови, холестерина, триглицеридов, липопротеидов низкой и высокой плотности (исследование уровня липопротеинов в крови), щелочной фосфатазы, креатинина, мочевины, мочевой кислоты, аммония, аланинаминотрансферазы (АЛТ), аспартатаминотрансферазы (АСТ), гаммаглютамилтрансферазы (ГГТ), креатинфосфокиназы (КФК), Исследование уровня/активности изоферментов креатинкиназы в крови, лактатдегидрогеназа (ЛДГ), кальция общего и ионизированного, натрия, калия, неорганического фосфора, железа, ферритина, магния, хлора, молочной кислоты), инсулина, показателей кислотно-основного равновесия (Исследование уровня буферных веществ в крови, Исследование уровня водородных ионов (рН) крови), исследование уровня молочной кислоты в крови (лактата) с целью оценки состояния печени, почек, баланса важнейших нутриентов при диетотерапии, кальциево-фосфорного обмена а также для оценки риска метаболической декомпенсации [12, 132].
- Рекомендуется определение лабораторных показателей (исследование кислотно-основного состояния и газов крови(Исследование уровня буферных веществ в крови, Исследование уровня водородных ионов (рН) крови), исследование уровня молочной кислоты в крови (лактата), уровня глюкозы в крови, уровня калия в крови, уровня натрия в крови, уровня ионизированного кальция в крови, уровня хлоридов в крови) в период и при подозрении на метаболический криз пациентам с клиническими и/или биохимическими признаками FAOD с целью оценки состояния и своевременной коррекции терапии [12, 132].
- Рекомендовано всем пациентам с клиническими и/или биохимическими признаками FAOD общий (клинический) анализ мочи, определение кетоновых тел в моче для оценки состояния мочевыводящих путей и почек [1, 2, 5].
- Рекомендуется всем пациентам с патологией сердечно-сосудистой системы исследования уровня N-терминального фрагмента натрийуретического пропептида мозгового (NT-proBNP) в крови для своевременной диагностики сердечной недостаточности, дифференциальной диагностики с одышкой, вызванной респираторными нарушениями, для решения вопросов о старте/коррекции кардиотропной терапии [11, 133].
- Рекомендовано всем пациентам с клиническими и/или биохимическими признаками FAOD регистрация электрогардиограммы и проведение ЭхоКГ для оценки состояния сердца, при наличии нарушении ритма и проводимости проведение холтеровского мониторирования сердечного ритма [11, 97, 109, 129].
- Рекомендовано всем пациентам с клиническими и/или биохимическими признаками FAOD ультразвукового исследования (УЗИ) органов брюшной полости и почек для исключения/подтверждения патологии печени и почек [9, 12, 13, 15, 145].
- Рекомендовано всем пациентам с клиническими и/или биохимическими признаками FAOD проведение офтальмоскопии с целью выявления пигментной дегенерации сетчатки [1, 2, 5].
- Рекомендуются при постановке диагноза пациентам с клиническими и/или биохимическими признаками FAOD применять мультидисциплинарный подход в виду того, что заболевания характеризуются поражением многих органов и систем, требует комплексной терапии, что диктует необходимость совместного ведения пациента специалистами разных профилей [12,13, 15, 145].
Лечение
Лечение, включая медикаментозную и немедикаментозную терапии, диетотерапию, обезболивание, медицинские показания и противопоказания к применению методов лечения
- Рекомендуется избегать длительного голодания всем пациентам с FAOD для предотвращения развития метаболических кризов [11-14, 145].
Начиная с 8–12 месяцев, с целью предотвращения катаболизма в ночной период, можно давать перед сном кукурузный крахмал (1 г/ кг /день (разведение крахмала в детской смеси или в воде 1:2), при регистрации гипогликемии в ночные часы возможно дополнительное введение крахмала в той же дозе) для обеспечения поступления адекватной энергии в течение ночи, данная тактика обычно используется только у пациентов с тяжелым течением заболевания, склонных к гипогликемии. В приложении А9 приведены среднесуточные нормы физиологических потребностей в энергии для детей.
- Рекомендовано назначение диетотерапии всем пациентам с FAOD (кроме MCADD, SCADD, системной недостаточности карнитина) после установления точного диагноза с целью снижения рисков возникновения метаболической декомпенсации [11-14].
Лечение пациентов с FAOD в период интеркуррентных инфекций
- Рекомендуется поддерживать высокое потребление углеводов во время любого метаболического стресса всем пациентам с FAOD для предотвращения развития метаболического криза [110].
Рекомендации по лечению пациентов с FAOD в период метаболической декомпенсации
- Рекомендуется внутривенное введение раствора декстрозы** под контролем ее уровня в крови пациентам с FAOD при развитии метаболического криза [5, 12 ,52, 130, 150].
- Рекомендуется коррекция метаболического ацидоза (при уровне бикарбонатов сыворотки крови <16 мЭкв/л) пациентам с FAOD при развитии метаболического криза [5, 12, 52].
- Рекомендуется коррекция водно-электролитных нарушений пациентам с FAOD при развитии метаболического криза [5,12, 145].
- Рекомендовано: назначение натрия бензоата (биологически активная добавка) при уровне аммиака в крови выше 150-200 мкмоль/л пациентам с FAOD при развитии метаболического криза [152, 154].
- Рекомендуется коррекция диетических мероприятий всем пациентам с FAOD в период метаболического криза с целью компенсации данного состояния [1, 105].
- Рекомендуется использовать приемлемый энтеральный способ кормления для пациента с FAOD для предотвращения развития катаболических состояний [1, 105].
- В случае необходимости введения эпинефрина** всем пациентам с FAOD рекомендовано его введение вместе с 10% раствором декстрозы** [97, 151].
2. Рекомендации по терапии отдельных форм FAOD
Рекомендации по лечению системной недостаточности карнитина
- Не рекомендовано назначение специальной диетотерапии пациентам с системной недостаточностью карнитина, поскольку ведущим патогенетическим механизмом развития заболевания является дефицит карнитина [82].
- Рекомендовано назначение #левокарнитина в дозе 50–400 мг/кг/сутки, всем пациентам с подтвержденной первичной недостаточностью карнитина с целью снижения риска возникновения гипогликемических эпизодов [54, 82, 155].
Рекомендации по лечению дефицита карнитин-ацилкарнитин транслоказы
- Рекомендуется пациентам с установленным диагнозом СAСT соблюдение низкожировой диеты (потребление жиров должно составлять 30% или менее от общей калорийности, 20% из них - MCT с низким содержанием C10 и 10% LCT (длинноцепочечные триглицериды) для предотвращения развития метаболического криза [66, 87-89].
- Рекомендуется пациентам с установленным диагнозом СAСT избегать длительных перерывов в приеме пищи для предотвращения гипогликемических состояний [66, 87-89].
- Рекомендуются пациентам с установленным диагнозом СAСT добавки незаменимых жирных кислот, с целью предотвращения их дефицита - линолевой (3–4%) и линоленовой (0,5–1%) - в соотношении от 5:1 до 10:1, их источником могут являются масло грецкого ореха, льняное, соевое [66, 87-89].
- Рекомендуется пациентам с установленным диагнозом СAСT в период интекуррентных инфекций и других состояний, при которых активируются механизмы катаболизма, применение перорально растворов декстрозы** или полимеров декстрозы в виде пищевой добавки (мальтодекстрин (декстринмальтоза)) и повышенное потребление углеводов [89].
- Рекомендуется пациентам с установленным диагнозом CPT 2 и CPT 1 избегать длительных перерывов в приеме пищи для предотвращения гипогликемических состояний. [66].
- Рекомендуется диета с высоким содержанием углеводов (70% калорий) с низким содержанием жира (<20% калорий) пациентам с установленным диагнозом дефицит CPT 2 и CPT 1 для предотвращения гипогликемических состояний и метаболической декомпенсации [91].
- Рекомендуется в период интеркуррентных инфекций и других состояний, при которых активируются механизмы катаболизма, пациентам с установленным диагнозом дефицит CPT 2 и CPT 1 с целью предотвращения метаболической декомпенсации употреблять примерно треть от общего количества калорий в виде МСТ [91].
- С целью профилактики эпизодов миоглобинурии пациентам с установленным диагнозом CPT 1, CPT2 рекомендуется избегать физических нагрузок во время интеркуррентных инфекций и при длительных перерывах в приеме пищи [91, 92].
- Взрослым пациентам с установленным диагнозом CPT 1, CPT2 в период метаболического криза, сопровождаемого рабдомиолизом и миоглобинурией рекомендуется применение стандартного протокола лечения этого осложнения, чтобы предотвратить острую почечную недостаточность [13, 92, 93].
Рекомендации по рациону питания при дефиците SCAD
Рекомендации по рациону питания при дефиците MCAD
- Рекомендуется пациентам с установленным диагнозом на недостаточность MCAD прием левокарнитина в дозе 60-100 мг на кг в сутки для коррекции вторичного дефицита с целью усиления выведения токсичных метаболитов [130, 149].
- Рекомендуются соблюдать интервалы между кормлениями не более 3-4 часов для доношенных детей с выявленными изменениями в анализе крови при проведении неонатального скрининга, характерными для дефицита MCAD, пока диагноз не будет подтвержден или исключен на основании дополнительных тестов [98].
- Рекомендуется соблюдать «безопасные» интервалы между кормлением для новорожденных с установленным диагнозом MCAD для предотвращения эпизодов метаболической декомпенсации (Приложение А9) [98].
- Рекомендуется новорожденным с установленным диагнозом недостаточность MCAD при грудном вскармливании дополнить питание адаптированной молочной смесью в течение первых трех полных дней (72 часа) для достижения необходимой потребности в калориях [98].
Рекомендации по диетотерапии пациентов с MCAD
- Рекомендуется всем пациентам с установленным диагнозом недостаточность MCAD соблюдать интервалы между кормлениями для предотвращения развития гипогликемических состояний (Приложение А8) [98, 94].
- Рекомендуется младенцам первого года жизни с установленным диагнозом недостаточность MCAD кормление материнским молоком или молочными смесями без ограничения LCF и без добавок МСТ. Рекомендуется соблюдать интервалы между кормлениями для предотвращения развития гипогликемических состояний (Приложение А8) [98].
- Рекомендовано у взрослых пациентов при дефиците MCAD избегать длительного голодания для предотвращения гипогликемических состояний и метаболических кризов [94, 98].
- Рекомендовано при наличии у пациента с недостаточностью MCAD признаков рекуррентного заболевания, выпаивание растворами, содержащими полимер декстрозы, чтобы предотвратить метаболическую декомпенсацию [94, 98].
- Рекомендовано при наличии у пациента с недостаточностью MCAD в период метаболической декомпенсации в случае сложностей приема растворов через рот проведение внутривенной инфузионной терапии растворами, содержащим декстрозу** [94, 98].
- Не рекомендовано пациентам с недостаточностью MCAD в период метаболической декомпенсации введение жиров с целью предотвращения усугубления тяжести состояния [98, 99].
- Рекомендуется всем взрослым пациентам с установленным диагнозом недостаточность MCAD избегать диеты с высоким содержанием жиров, употребление алкоголя, что вызывает метаболическую декомпенсацию [94, 99].
- Рекомендуется избегать применения ацетилсалициловой кислоты** всем пациентам с установленным диагнозом MCAD с целью предотвращения развития метаболического криза и синдрома Рейе [94, 100].
Рекомендации по рациону питания при дефиците LCHAD и TFP
- Рекомендовано у пациентов при дефиците LCHAD и TFP придерживаться максимально низкого потребление ДЦТ, как при наличии симптомов, так и при их отсутствии, для профилактики возникновения полинейропатии [66].
- Рекомендуется назначение диетотерапии при выявлении характерных изменений по результатам неонатального скрининга и/или биохимической диагностики, не дожидаясь подтверждения диагноза молекулярно-генетическим методом [66].
- Рекомендуется при введении твердой пищи в рацион пациентов с недостаточностью LCHAD и TFP поддерживать содержание общих жиров в рационе 25–30% от общего потребления калорий, где 20–25% приходится на MCT и 5–10% - на LCT (Приложение А14) [66].
Рекомендации по рациону питания при дефиците VLCAD
- Рекомендуется продолжать кормление грудным молоком или адаптированной детской смесью с добавлением специализированной низкожировой смеси с ограничением LCT из расчета 50/50 пациентам с бессимптомным вариантом VLCAD, если результаты стандартных лабораторных исследований, таких как определение активности креатинфосфокиназы в крови, активности аланинамитрансферазы в крови, аспартатаминотрансферазы в крови, глюкозы в крови находятся в пределах нормы [66].
- Рекомендовано младенцам и более взрослым детям с бессимптомным вариантом VLCAD снижение содержания жиров в рационе до 30–40% от общей калорийности из них 10-15% энергии за счет МСТ с целью предотвращения развития метаболического криза [66].
- Рекомендуется при наличии симптомов у пациентов с дефицитом VLCAD диета с пониженным содержанием LCFA и добавление MCT жиров для предотвращения рабдомиолиза и метболической декомпенсации [61, 66].
Рекомендации по лечению пациентов с ГА2
- Рекомендуется пациентам с установленным диагнозом ГА2 соблюдение диеты с низким содержанием жиров (20-25% энергии), низким содержанием белка для снижения избыточного потребления изолейцина, лейцина, лизина, триптофана и валина [103, 111].
- Не рекомендуется пациентам с установленным диагнозом ГА2 назначение МСТ [104].
- Рекомендуется во время кризов пациентам с установленным диагнозом ГА2 парентеральное введение декстрозы** для купирования гипогликемии и подавления липолизиса, натрия бензоат (биологически активная добавка) для купирования гипераммониемии [111].
3. Рекомендации по применению отдельных лекарственных препаратов
Рекомендации по применению отдельных лекарственных препаратов, содержащих левокарнитин
- Рекомендуется избегать назначения Левокарнитина в период криза при дефектах окисления длинноцепочечных жирных кислот, особенно в виде внутривенных инфузий для предотвращения развития рабдомиолиза, неврологических нарушений и аритмии [63, 142, 132].
- Рекомендуется пациентам с недостаточностью МТР и LCHAD, назначать биологическую активную добавку в виде докозагексаеновой кислоты (DHA), играющей важную роль в формировании функций нервной и иммунной систем, а также зрительного анализатора, в дозировке 60 -65 мг/день детям с весом тела <20 кг и в дозировке 130мг в день – детям с весом тела >20 кг для поддержания функций нервной и иммунной системы [105, 137, 157].
- В межприступный период для пациентов с FAOD находящихся на диете с ограничением длинноцепочечных жиров рекомендовано добавлять натуральные растительные масла (кокосовое масло, масло грецкого ореха) для восполнения недостатка жирных кислот [105].
- Рекомендовано назначение незаменимых жирных кислот пациентам с FAOD на фоне низкожировой диетотерапии с целью компенсации их недостаточности [11-14, 105].
Рекомендации по применению среднецепочечных триглицеридов
- Рекомендовано в качестве основных пищевых источников среднецепочечных жирных кислот использовать 50% эмульсию MCT в питании пациентов с недостаточности TFP и LCHAD, и VLCAD для предотвращения развития мышечной симптоматики [112, 149].
- Не рекомендуется назначение пациентам с MCAD триглицеридов со средней длиной цепи (MCT) с целью предотвращения ухудшения состояния пациента. развития метаболического криза [105].
- Рекомендуется пациентам с FAOD назначение витаминов группы В и жирорастворимых витаминов А, D, Е в возрастных дозировках с целью предотвращения их дефицита [5,12, 105].Уровень убедительности доказательств C (уровень достоверности рекомендации – 5).
- Рекомендуется всем пациентам с биохимическими нарушениями, характерными для ГА2 назначение (биологической активной добавки) рибофлавина [106, 111, 135, 136].Уровень убедительности доказательств C (уровень достоверности рекомендации – 5).
4. Симптоматическое лечение
- Не рекомендуется применять препараты вальпроевой кислоты** в случае необходимости назначения противоэпилептической терапии с целью предотвращения развития печеночной недостаточности [5,12, 145].
5. Хирургическое лечение
Медицинская реабилитация
Медицинская реабилитация и санаторно-курортное лечение, медицинские показания и противопоказания к применению методов медицинской реабилитации, в том числе основанных на использовании природных лечебных факторов
- Рекомендуется пациентам и членам их семей проводить консультацию/консультации медицинского психолога с целью повышения приверженности к лечению [1, 2, 14].
Госпитализация
Организация оказания медицинской помощи
Показания для плановой госпитализации:
- необходимость проведения различных видов экспертиз или обследования в медицинской организации при невозможности проведения их в амбулаторных условиях, требующих динамического наблюдения (в том числе оформление заключения федерального консилиума).
Показания для экстренной госпитализации:
- острые заболевания;
- обострения хронических болезней;
- отравления и травмы, состояния, требующие интенсивной терапии и перевода в реанимационные отделения или отделения интенсивной терапии а также круглосуточного медицинского наблюдении и проведения специальных видов обследования и лечения.
Показания к выписке пациента из медицинской организации:
- отсутствие угрозы жизни больного;
- отсутствие угрозы развития осложнений, требующих неотложного лечения;
- стабилизация состояния и основных клинико-лабораторных показателей патологического процесса по основному заболеванию;
- отсутствие необходимости в постоянном врачебном и круглосуточном медицинском наблюдении по основному заболеванию;
- необходимости перевода больного в другую больницу или учреждение социального обеспечения.
Пациентам с FAOD 1 раз в 6-12 мес. (в соответствии с тяжестью состояния) показано комплексное обследование в многопрофильных стационарах.
Продолжительность госпитализации зависит от скорости коррекции метаболических нарушений, а также от сроков появления положительной динамики со стороны центральной нервной системы и других органов, скорости восстановления показателей глюкозы крови и кислотно-щелочного состояния, ответ на лечение отмечается в течение 5-7 дней. Пребывание в стационаре в среднем составляет 21 день.
После выписки из стационара дети нуждаются в наблюдении педиатра, невролога, кардиолога, гастроэнтеролога, офтальмолога, диетолога, генетика, проведении ЭКГ (по показаниям).
Наблюдение пациентов по месту жительства (в амбулаторно-поликлинических условиях) должно осуществляться постоянно.
Профилактика
Профилактика и диспансерное наблюдение, медицинские показания и противопоказания к применению методов профилактики
1. Профилактика
- Рекомендуется медико-генетическое консультирование после установления диагноза пациентам с FAOD, с целью разъяснений генетического риска, возможности проведения пренатальной и преимплантационной диагностики для любой последующей беременности в семьях, отягощенных хотя бы одним случаем FAOD [1,2, 6].
- Рекомендуется консультация женщин-носительниц FAOD для разъяснения рисков возникновения акушерских осложнений [105, 138].
2. Диспансерное наблюдение
- Рекомендуется проведение исследования уровня свободного карнитина и ацилкарнитинов в крови у пациентов с FAOD с целью контроля уровня свободного карнитина и ацилкарнитинов [19, 21].
- Рекомендовано всем пациентам с диагнозом FAOD общий (клинический) анализ крови развернутый (гемоглобин, количество эритроцитов, цветовой показатель, количество лейкоцитов, тромбоцитов, лейкоцитарная формула и скорость оседания эритроцитов) для оценки основных параметров кроветворения и наличия воспалительных процессов [1, 2, 5, 28].
- Рекомендуется всем пациентам с FAOD проведение анализа крови биохимического общетерапевтического (содержание глюкозы, общего белка, белковых фракций, альбумин, С-реактивного белка, общего билирубина и его фракции, холестерина, триглицеридов, липопротеидов низкой и высокой плотности (исследование уровня липопротеинов в крови), щелочной фосфатазы, креатинина, мочевины, мочевой кислоты, аммония, аланинаминотрансферазы (АЛТ), аспартатаминотрансферазы (АСТ), гаммаглютамилтрансферазы (ГГТ), креатинфосфокиназы (КФК), Исследование уровня/активности изоферментов креатинкиназы в крови, лактатдегидрогеназа (ЛДГ), кальция общего и ионизированного, натрия, калия, неорганического фосфора, железа, ферритина, магния, хлора, молочной кислоты), уровня инсулина плазмы крови, исследование кислотно-основного состояния и газов крови (Исследование уровня буферных веществ в крови, Исследование уровня водородных ионов (рН) крови), исследование уровня молочной кислоты в крови (лактата) с целью оценки состояния печени, почек, баланса важнейших нутриентов при диетотерапии, кальциево-фосфорного обмена и выявления отклонений важных биохимических показателей для дальнейшей коррекции [12, 132, 145].
- Рекомендовано всем пациентам с FAOD общий (клинический) анализ мочи, обнаружние кетоновых тел в моче для оценки состояния мочевыводящих путей и почек [1, 2, 5, 34].
- Рекомендуется определение лабораторных показателей (исследование кислотно-основного состояния газов крови (Исследование уровня буферных веществ в крови, Исследование уровня водородных ионов (рН) крови), исследование уровня молочной кислоты в крови (лактата), глюкоза, электролиты ( исследование уровня калия, натрия, ионизированного кальция, хлоридов в крови) в период и при подозрении на метаболический криз пациентам с клиническими и/или биохимическими признаками FAOD с целью оценки состояния и своевременной коррекции терапии [12, 132].
- Рекомендуется всем пациентам с патологией сердечно-сосудистой системы исследования уровня N-терминального фрагмента натрийуретического пропептида мозгового (NT-proBNP) в крови для своевременной диагностики сердечной недостаточности, дифференциальной диагностики с одышкой, вызванной респираторными нарушениями, для решения вопросов о старте/коррекции кардиотропной терапии [11, 133].
- Рекомендуется проведение холтеровское мониторирование сердечного ритма, Эхо-КГ, пациентам с FAOD при постановке диагноза, далее не реже 1 раза в год с целью диагностики патологии со стороны ССС [97, 109, 129, 133].
- Рекомендуется проведение электроэнцефалографии (ЭЭГ) пациентам с FAOD при наличии неврологической симптоматики для выявления эпилептической активности и для контроля противоэпилептической терапии согласно клиническим рекомендациям по эпилепсии [35, 158].
- Рекомендуется проведение ультразвукового исследования (УЗИ) органов брюшной полости пациентам с FAOD для выявления патологии печени и почек [9, 12,13, 145].
- Рекомендуется проведение офтальмоскопии пациентам с FAOD с целью своевременного обнаружения офтальмологической патологии [38, 149].
- Рекомендуется пациентам с FAOD проведение коррекции лечебного питания и симптоматической терапии с целью исключения белково-энергетической недостаточности [11-14, 105].
- Рекомендуются для наблюдения пациентов с установленным диагнозом FAOD применять мультидисциплинарный подход в виду того, что заболевания характеризуются поражением многих органов и систем, требует комплексной терапии, что диктует необходимость совместного ведения пациента специалистами разных профилей [12,13, 15, 28, 145].
- Рекомендуется обучение родителей (законных представителей) /пациентов правилам организации терапии в межприступный период и в период угрозы метаболического криза пациентов с FAOD с целью предотвращения развития повторных метаболических кризов [105].
Информация
Источники и литература
-
Клинические рекомендации Российского общества медицинских генетиков
- Клинические рекомендации Российского общества медицинских генетиков - 1. Михайлова СВ, Захарова ЕЮ, Петрухин АС. Нейрометаболические заболевания у детей и подростков: диагностика и подходы к лечению (2-е изд., переработанное и дополненное) / М.: Литтерра, 2017. 2. Orngreen MC, Nørgaard MG, Sacchetti M, van Engelen BG, Vissing J. Fuel utilization in patients with very long-chain acyl-coa dehydrogenase deficiency. Ann Neurol. 2004 Aug;56(2):279-83 3. Краснопольская КД Наследственные болезни обмена веществ. Справочное пособие для врачей. /М., 2005. С. 43-51. 4. Andresen BS, Olpin S, Poorthuis BJ et al (1999) Clear correlation of genotype with disease phenotype In very-long-chain acyl-CoA dehydrogenase deficiency. Am J Hum Genet 64:479–494; 5. Gregersen N, Andresen BS, Corydon MJ, Corydon TJ, Olsen RK, Bolund L, Bross P. Mutation analysis in mitochondrial fatty acid oxidation defects: Exemplified by acyl-CoA dehydrogenase deficiencies, with special focus on genotype-phenotype relationship. Hum Mutat. 2001 Sep;18(3):169-89; 6. Spiekerkoetter U, Khuchua Z, Yue Z, Bennett MJ, Strauss AW. General mitochondrial trifunctional protein (TFP) deficiency as a result of either alpha- or beta-subunit mutations exhibits similar phenotypes because mutations in either subunit alter TFP complex expression and subunit turnover. Pediatric Research. 2004;55(2):190–196. doi:10.1203/01.PDR.0000103931.80055.06 7. Wilcken B.Recent advances in newborn screening. J Inherit Metab Dis. 2007 Apr;30(2):129-33. 8. Wilcken B. Fatty acid oxidation disorders: outcome and long-term prognosis. J Inherit Metab Dis. 2010;33(5):501–506 9. Baruteau J, Sachs P, Broué P, Brivet M, Abdoul H, Vianey-Saban C, Ogier de Baulny H. Clinical and biological features at diagnosis in mitochondrial fatty acid beta-oxidation defects: a French pediatric study of 187 patients. J Inherit Metab Dis. 2013 Sep;36(5):795-803 10. Saudubray JM, et al. Recognition and management of fatty acid oxidation defects: a series of 107 patients. J Inherit Metab Dis. 1999;22(4):488–502. 11. Bonnet D, et al. Arrhythmias and conduction defects as presenting symptoms of fatty acid oxidation disorders in children. Circulation. 1999;100(22):2248–2253. 12. Wanders R.J.A., Vreken P., den Boer M.E.J. et al. Disorders of mitochondrial fatty acyl-CoA β-oxidation. J Inher Metab Dis 1999; 22: 442—487. 13. Николаева Е.А., Мамедов И.С. «Диагностика наследственных дефектов обмена жирных кислот у детей» Российский вестник перинатологии и педиатрии, 2008: 6:66–8014. Николаева Е.А., Леонтьева И.В., Калачанова Е.П., Золкина И.В. Задержка физического развития и кардиомиопатия у ребенка с первичным системным дефицитом карнитина. Трудный пациент 2012; 2-3: 50-54. 15. МP 2.3.1.2432-08 "Нормы физиологических потребностей в энергии и пищевых веществах для различных групп населения Российской Федерации" (утв. Главным государственным санитарным врачом РФ 18 декабря 2008 г.). 16. Wilcken B, et al. Pregnancy and fetal long-chain 3-hydroxyacyl coenzyme a dehydrogenase deficiency. Lancet. 1993;341(8842):407–408 17. Makhseed N, et al. Carnitine transporter defect due to a novel mutation in the SLC22A5 gene presenting with peripheral neuropathy. J Inherit Metab Dis. 2004;27(6):778–780., 18. Wang Y, et al. Phenotype and genotype variation in primary carnitine deficiency. Genet Med. 2001;3(6):387–392. 19. Innes AM, et al. Hepatic carnitine palmitoyltransferase I deficiency presenting as maternal illness in pregnancy. Pediatr Res. 2000;47(1):43–45 20. Al-Thihli K, Sinclair G, Sirrs S, Mezei M, Nelson J, Vallance H. Performance of serum and dried blood spot acylcarnitine profiles for detection of fatty acid β-oxidation disorders in adult patients with rhabdomyolysis. J Inherit Metab Dis. 2014. Mar;37(2):207-13. 21. Arnold GL, et al. A Delphi clinical practice protocol for the management of very long chain acyl-CoA dehydrogenase deficiency. Mol Genet Metab. 2009;96(3):85–90 22. Bach AC, Babayan VK. Medium-chain triglycerides: an update. Am J Clin Nutr. 1982;36(5):950– 962. doi: 10.1093/ajcn/36.5.950. 23. Bartlett K, Eaton S. Mitochondrial β-oxidation. Eur J Biochem. 2004;271(3):462–469. 24. Baruteau J, et al. Clinical and biological features at diagnosis in mitochondrial fatty acid beta-oxidation defects: a French pediatric study of 187 patients. J Inherit Metab Dis. 2013;36(5):795– 803. 25. Baruteau J, et al. Clinical and biological features at diagnosis in mitochondrial fatty acid beta-oxidation defects: a French pediatric study from 187 patients. Complementary data. J Inherit Metab Dis. 2014;37(1):137–139. 26. Bastin J, Lopes-Costa A, Djouadi F. Exposure to resveratrol triggers pharmacological correction of fatty acid utilization in human fatty acid oxidation-deficient fibroblasts. Hum Mol Genet. 2011;20(10):2048–2057. 27. Berardo A, DiMauro S, Hirano M. A diagnostic algorithm for metabolic myopathies. Curr Neurol Neurosci Rep. 2010;10:118–26.28. Bleeker JC, Houtkooper RH. Sirtuin activation as a therapeutic approach against inborn errors of metabolism. J Inherit Metab Dis. 2016;39:565–572. 29. Bleeker JC, Kok IL, Ferdinandusse S, et al. Proposal for an individualized dietary strategy in patients with very long-chain acyl-CoA dehydrogenase deficiency. J Inherit Metab Dis 2018. 30. Bonnefont JP, et al. Long-term follow-up of bezafibrate treatment in patients with the myopathic form of carnitine palmitoyltransferase 2 deficiency. Clin Pharmacol Ther. 2010;88(1):101–108. 31. Borch L, et al. Normal levels of plasma free carnitine and Acylcarnitines in follow-up samples from a Presymptomatic case of carnitine Palmitoyl transferase 1 (CPT1) deficiency detected through newborn screening in Denmark. JIMD Rep. 2012;3:11–15. 32. Burrage LC, et al. Elevations of C14:1 and C14:2 plasma Acylcarnitines in fasted children: a diagnostic dilemma. J Pediatr. 2016;169:208–213.e2. 33. Chegary M, et al. Mitochondrial long chain fatty acid beta-oxidation in man and mouse. Biochim Biophys Acta. 2009;1791(8):806–815. 34. Cox GF, et al. Reversal of severe hypertrophic cardiomyopathy and excellent neuropsychologic outcome in very-long-chain acyl-coenzyme a dehydrogenase deficiency. J Pediatr. 1998;133(2):247–253. 35. Cox PJ, et al. Nutritional ketosis alters fuel preference and thereby endurance performance in athletes. Cell Metab. 2016;24(2):256–268. 36. De Biase I, et al. Diagnosis, treatment, and clinical outcome of patients with mitochondrial trifunctional protein/long-chain 3-Hydroxy acyl-CoA dehydrogenase deficiency. JIMD Rep. 2017;31:63–71. doi: 10.1007/8904_2016_558. 37. Demaugre F, et al. Hepatic and muscular presentations of carnitine palmitoyl transferase deficiency: two distinct entities. Pediatr Res. 1988;24(3):308–311. 38. Deschauer M, Wieser T, Zierz S. Muscle carnitine palmitoyltransferase II deficiency: Clinical and molecular genetic features and diagnostic aspects. Arch Neurol. 2005;62:37– 41. 39. Diekman E, et al. The newborn screening paradox: sensitivity vs. Overdiagnosis in VLCAD deficiency. JIMD Rep. 2016;27:101–106. 40. Diekman EF, Ferdinandusse S, van der Pol L, Waterham HR, Ruiter JP, Ijlst L, Wanders RJ, Houten SM, Wijburg FA, Blank AC, Asselbergs FW, Houtkooper RH, Visser G. Fatty acid oxidation flux predicts the clinical severity of VLCAD deficiency. Genet Med. 2015 Apr 2. 41. Diekman EF, van Weeghel M, Wanders RJ, Visser G, Houten SM. Food withdrawal lowers energy expenditure and induces inactivity in long-chain fatty acid oxidation-deficient mouse models. FASEB J. 2014 Jul;28(7):2891-900.42. Djouadi F, et al. Bezafibrate increases very-long-chain acyl-CoA dehydrogenase protein and mRNA expression in deficient fibroblasts and is a potential therapy for fatty acid oxidation disorders. Hum Mol Genet. 2005;14(18):2695–2703. 43. Djouadi F, et al. Peroxisome proliferator activated receptor delta (PPARdelta) agonist but not PPARalpha corrects carnitine palmitoyl transferase 2 deficiency in human muscle cells. J Clin Endocrinol Metab. 2005;90(3):1791–1797. 44. Elsayed EF, Reilly RF. Rhabdomyolysis: A review, with emphasis on the pediatric population. Pediatr Nephrol.2010;25:7–18. 45. Erguven Muferet, Yılmaz Oznur, Koc Seher, Cakı Suar, Ayhan Yusuf, Donmez Metin, Dolunay Gulderen. A Case of Early Diagnosed Carnitine Deficiency Presenting with Respiratory Symptoms. Annals of Nutrition and Metabolism. 2007;51(4):331–334. doi: 10.1159/000107675. 46. Evans M, et al. VLCAD deficiency: follow-up and outcome of patients diagnosed through newborn screening in Victoria. Mol Genet Metab. 2016;118(4):282–287. 47. Gillingham MB, et al. Triheptanoin versus trioctanoin for long-chain fatty acid oxidation disorders: a double blinded, randomized controlled trial. J Inherit Metab Dis. 2017;40(6):831– 843. 48. Houten SM, Violante S, Ventura FV, Wanders RJA. The biochemistry and physiology of mitochondrial fatty acid beta-oxidation and its genetic disorders. Annu Rev Physiol. 2016;78:23– 44. 49. Ibdah JA. Acute fatty liver of pregnancy: an update on pathogenesis and clinical implications. World J Gastroenterol. 2006;12(46):7397–7404. 50. IJlst L, et al. Common missense mutation G1528C in long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency. Characterization and expression of the mutant protein, mutation analysis on genomic DNA and chromosomal localization of the mitochondrial trifunctional protein alpha subunit gene. J Clin Invest. 1996;98(4):1028–1033. 51. Kilfoyle D, Hutchinson D, Potter H, George P. Recurrent myoglobinuria due to carnitine palmitoyltransferase II deficiency: Clinical, biochemical, and genetic features of adult-onset cases. N Z Med J. 2005;118:U1320. 52. Kluge Stefan, Kühnelt Peter, Block Andreas, Merkel Martin, Gocht Andreas, Lukacs Zoltan, Kohlschütter Alfried, Kreymann Georg. A young woman with persistent hypoglycemia, rhabdomyolysis, and coma: Recognizing fatty acid oxidation defects in adults. Critical Care Medicine. 2003;31(4):1273–1276. 53. Liu J, Ghaziani TT, Wolf JL. Acute fatty liver disease of pregnancy: updates in pathogenesis, diagnosis, and management. Am J Gastroenterol. 2017;112(6):838–846.54. Longo N, Amat di San Filippo C, Pasquali M. Disorders of carnitine transport and the carnitine cycle. Am J Med Genet C Semin Med Genet. 2006;142c(2):77–85. doi: 10.1002/ajmg.c.30087 55. Lopaschuk GD, et al. Myocardial fatty acid metabolism in health and disease. Physiol Rev. 2010;90(1):207–258.
- Клинические рекомендации Российского общества медицинских генетиков - 56. Moore SJ, Haites NE, Broom I, White I, Coleman RJ, Pourfarzam M, et al. Acylcarnitine analysis in the investigation of myopathy. J Inherit Metab Dis. 1998;21:427–8. 57. Moorthie S, et al. Systematic review and meta-analysis to estimate the birth prevalence of five inherited metabolic diseases. J Inherit Metab Dis. 2014;37(6):889–898. 58. Oliveira SF, Pinho L, Rocha H, Nogueira C, Vilarinho L, Dinis MJ, Silva C. Rhabdomyolysis as a presenting manifestation of very long-chain acyl-coenzyme a dehydrogenase deficiency. Clin Pract. 2013 Aug 6;3(2):e22. 59. Rashed MS, et al. Diagnosis of inborn errors of metabolism from blood spots by acylcarnitines and amino acids profiling using automated electrospray tandem mass spectrometry. Pediatr Res. 1995;38(3):324–331. 60. Saudubray JM, et al. Recognition and management of fatty acid oxidation defects: a series of 107 patients. J Inherit Metab Dis. 1999;22(4):488–502. doi: 10.1023/A:1005556207210. 61. Sharef Waadallah Sharef, Khalfan Al-Senaidi, and Surendra Nath Joshi. Successful Treatment of Cardiomyopathy due to Very Long-Chain Acyl-CoA Dehydrogenase Deficiency: First Case Report from Oman with Literature Review. Oman Medical Journal (2013) Vol. 28, No. 5:354-356. 62. Solis JO, Singh RH. Management of fatty acid oxidation disorders: A survey of current treatment strategies. J Am Diet Assoc. 2002;102:1800–3. 63. Spiekerkoetter U,Bastin J., Gillingham M., Morris A // Current issues regarding treatment of mitochondrial fatty acid oxidation disorders. J Inherit Metab Dis . 2010 Oct;33(5):555-61. doi: 10.1007/s10545-010-9188-1. 64. Spiekerkoetter U, et al. Lethal undiagnosed very long-chain acyl-CoA dehydrogenase deficiency with mild C14-Acylcarnitine abnormalities on newborn screening. JIMD Rep. 2012;6:113–115. 65. Spiekerkoetter U, et al. Management and outcome in 75 individuals with long-chain fatty acid oxidation defects: results from a workshop. J Inherit Metab Dis. 2009;32(4):488–497. doi: 10.1007/s10545-009-1125-9. 66. Spiekerkoetter U, Lindner M, Santer R, et al. Treatment recommendations in long-chain fatty acid oxidation defects: consensus from a workshop. J Inherit Metab Dis. 2009; 32:498–505. doi: 10.1007/s10545-009-1126-8 67. Spiekerkoetter U, Mayatepek E. Update on mitochondrial fatty acid oxidation disorders. J Inherit Metab Dis. 2010;33(5):467–468. doi: 10.1007/s10545-010-9208-1.68. Spiekerkoetter U. Mitochondrial fatty acid oxidation disorders: clinical presentation of long-chain fatty acid oxidation defects before and after newborn screening. J Inherit Metab Dis. 2010;33(5):527–532. doi: 10.1007/s10545-010-9090-x. 69. Tenopoulou M, Chen J, Bastin J, Bennett MJ, Ischiropoulos H, Doulias PT. Strategies for correcting very long chain acyl-CoA dehydrogenase deficiency. J Biol Chem. 2015 Apr 17;290(16):10486-94. 70. Topcu Y, et al. Importance of acylcarnitine profile analysis for disorders of lipid metabolism in adolescent patients with recurrent rhabdomyolysis: report of two cases. Ann Indian Acad Neurol. 2014;17(4):437–440. 71. Tucci S, Flögel U, Hermann S, Sturm M, Schäfers M, Spiekerkoetter U. Development and pathomechanisms of cardiomyopathy in very long-chain acyl-CoA dehydrogenase deficient (VLCAD (-/-)) mice. Biochim Biophys Acta. 2014 May;1842(5):677-85. 72. Wüst RC, et al. Ketones and inborn errors of metabolism: old friends revisited. J Inherit Metab Dis. 2017;40(1):3–4. 73. Xiong D, He H, James J, et al. Cardiac- specific VLCAD deficiency induces dilated cardiomyopathy and cold intolerance. Am J Physiol Heart Circ Physiol. 2014 Feb;306(3):H326-38. 74. Goetzman ES. Advances in the Understanding and Treatment of Mitochondrial Fatty Acid Oxidation Disorders. Curr Genet Med Rep. 2017;5(3):132–142. doi:10.1007/s40142-017-0125-6 75. Pedersen CB, Kolvraa S, Kolvraa A, et al. The ACADS gene variation spectrum in 114 patients with short-chain acyl-CoA dehydrogenase (SCAD) deficiency is dominated by missense variations leading to protein misfolding at the cellular level. Hum Genet 2008;124:43-56. 10.1007/s00439-008-0521-9 76. Wolfe L, Jethva R, Oglesbee D, et al. Short-Chain Acyl-CoA Dehydrogenase Deficiency. 2011 Sep 22 [Updated 2018 Aug 9]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2020. Available from: https://www.ncbi.nlm.nih.gov/books/NBK63582/https://www.ncbi.nlm.nih.gov/books/NBK63582/ 77. Martins E, Cardoso ML, Rodrigues E, et al. Short-chain 3-hydroxyacyl-CoA dehydrogenase deficiency: the clinical relevance of an early diagnosis and report of four new cases. J Inherit Metab Dis 2011;34:835-42. 10.1007/s10545-011-9287-7 78. Kapoor RR, James C, Flanagan SE, et al. 3-Hydroxyacyl-coenzyme A dehydrogenase deficiency and hyperinsulinemic hypoglycemia: characterization of a novel mutation and severe dietary protein sensitivity. J Clin Endocrinol Metab 2009;94:2221-5. 10.1210/jc.2009-0423 79. Clayton PT, Eaton S, Aynsley-Green A, et al. Hyperinsulinism in short-chain L-3-hydroxyacyl-CoA dehydrogenase deficiency reveals the importance of beta-oxidation in insulin secretion. JClin Invest 2001;108:457-65. 10.1172/JCI200111294 80. Tyni T, Ekholm E, Pihko H. Pregnancy complications are frequent in long-chain 3-hydroxyacyl-coenzyme a dehydrogenase deficiency. Am J Obstet Gynecol. 1998;178(3):603–608 81. «Nutrition Management of Inherited Metabolic Diseases» Laurie E. Bernstein, Fran Rohr, Joanna R. Helm 2015, Springer.com DOI 10.1007/978-3-319-14621-8 XIV 82. Magoulas, P.L., El-Hattab, A.W. Systemic primary carnitine deficiency: an overview of clinical manifestations, diagnosis, and management. Orphanet J Rare Dis 7, 68 (2012). https://doi.org/10.1186/1750-1172-7-68 83. Stanley CA, Bennett MJ, Longo N: Plasma membrane carnitine transporter defect. Online metabolic and molecular bases of inherited disease. Edited by Valle D, Beaudet AL, Vogelstein B, Kinzler KW, et al. 2006.http://www.ommbid.com/. Published January 2006. Updated March 28, 2011 84. Rubio-Gozalbo ME, Bakker JA, Waterham HR, Wanders RJ. Carnitine-acylcarnitine translocase deficiency, clinical, biochemical and genetic aspects. Mol Aspects Med. 2004;25:521–532. doi: 10.1016/j.mam.2004.06.007 85. Brivet M (2004) Carnitine-acylcarnitine translocase deficiency. Orphanet Encyclopedia 1–5 86. Lund AM, Skovby F, Vestergaard H, Christensen M, Christensen E. Clinical and biochemical monitoring of patients with fatty acid oxidation disorders. J Inherit Metab Dis. 2010;33:495–500. doi: 10.1007/s10545-009-9000-2 87. Vitoria I, Martín-Hernández E, Peña-Quintana L, et al. Carnitine-acylcarnitine translocase deficiency: experience with four cases in Spain and review of the literature. JIMD Rep. 2015;20:11–20. doi:10.1007/8904_2014_382 88. Iacobazzi V, Pasquali M, Singh R, et al. Response to therapy in carnitine/acylcarnitine translocase (CACT) deficiency due to a novel missense mutation. Am J Med Genet A. 2004;126A:150–155. doi: 10.1002/ajmg.a.20573 89. Spiekerkoetter U, Duran M. Mitochondrial fatty acid oxidation defects. In: Blau N, Duran M, Gibson KM, Dionisi-Visi C, editors. Physician’s guide to the diagnosis, treatment, and follow-up of inherited metabolic diseases. Berlin/Heidelberg: Springer-Verlag; 2014. pp. 247–264 90. Rubio-Gozalbo ME, Vos P, Forget PP, et al. Carnitine-acylcarnitine translocase deficiency: case report and review of the literature. Acta Paediatr. 2003;92:501–504. doi: 10.1111/j.1651-2227.2003.tb00586.x. 91. Bennett MJ, Santani AB. Carnitine Palmitoyltransferase 1A Deficiency. 2005 Jul 27 [Updated 2016 Mar 17]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet].Seattle (WA): University of Washington, Seattle; 1993-2020. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1527/https://www.ncbi.nlm.nih.gov/books/NBK1527/ 92. Angelini, C., Federico, A., Reichmann, H., Lombes, A., Chinnery, P. and Turnbull, D. (2006), Task force guidelines handbook: EFNS guidelines on diagnosis and management of fatty acid mitochondrial disorders. European Journal of Neurology, 13: 923-929. doi:10.1111/j.1468-1331.2006.01482.x 93. Better OS, Stein GH. Early management of shock and prophylaxis of acute renal failure in traumatic rhabdomyolisis. The New England Journal of Medicine 1990; 322: 825–82 94. Merritt JL 2nd, Chang IJ. Medium-Chain Acyl-Coenzyme A Dehydrogenase Deficiency. 2000 Apr 20 [Updated 2019 Jun 27]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2020. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1424/https://www.ncbi.nlm.nih.gov/books/NBK1424/ 95. Lee PJ, Harrison EL, Jones MG, Jones S, Leonard JV, Chalmers RA. L-carnitine and exercise tolerance in medium-chain acyl-coenzyme A dehydrogenase (MCAD) deficiency: a pilot study. J Inherit Metab Dis. 2005;28:141–52. 96. Madsen KL, Preisler N, Orngreen MC, Andersen SP, Olesen JH, Lund AM, Vissing J. Patients with medium-chain acyl-coenzyme a dehydrogenase deficiency have impaired oxidation of fat during exercise but no effect of L-carnitine supplementation. J Clin Endocrinol Metab. 2013;98:1667–75. 97. Saud H. Aldubayan, Lance H. Rodan, Gerard T. Berry,Harvey L. Levy. Acute Illness Protocol for Fatty Acid Oxidation and Carnitine Disorders. Pediatric Emergency Care, 2017. V33, Number 4, p 300. 98. (Clinical paediatric dietetics / edited by Vanessa Shaw. – Fourth edition. Copyright © 2015 John Wiley & Sons, Ltd, Print ISBN:9780470659984 |Online ISBN:9781118915349 |DOI:10.1002/9781118915349) 99. [ Lang TF. Adult presentations of medium-chain acyl-CoA dehydrogenase deficiency (MCADD). J Inherit Metab Dis. 2009;32:675–83]. 100. [ Uppala R, Dudiak B, Beck ME, Bharathi SS, Zhang Y, Stolz DB, Goetzman ES. Aspirin increases mitochondrial fatty acid oxidation. Biochem Biophys Res Commun. 2017;482:346–51]. 101. Fraser H, Geppert J, Johnson R, et al. Evaluation of earlier versus later dietary management in long-chain 3-hydroxyacyl-CoA dehydrogenase or mitochondrial trifunctional protein deficiency: a systematic review. Orphanet J Rare Dis. 2019;14(1):258. Published 2019 Nov 15. doi:10.1186/s13023-019-1226-y 102. Yamada, K., Taketani, T. Management and diagnosis of mitochondrial fatty acid oxidation disorders: focus on very-long-chain acyl-CoA dehydrogenase deficiency. J Hum Genet 64, 73–85(2019). https://doi.org/10.1038/s10038-018-0527-7 103. Angle B, Burton BK. Risk of sudden death and acute life-threatening events in patients with glutaric acidemia type II. Mol Genet Metab 2008;93:36-9. 10.1016/j.ymgme.2007.09.015] 104. [Gharbawy A, Vockley J. Inborn Errors of Metabolism with Myopathy: Defects of Fatty Acid Oxidation and the Carnitine Shuttle System. Pediatr Clin North Am 2018;65:317-35. 10.1016/j.pcl.2017.11.006].
- Клинические рекомендации Российского общества медицинских генетиков - 105. Merritt JL 2nd, Norris M, Kanungo S. Fatty acid oxidation disorders. Ann Transl Med. 2018;6(24):473. doi:10.21037/atm.2018.10.57. 106. Bosch AM, Abeling NG, Ijlst L, et al. Brown-Vialetto-Van Laere and Fazio Londe syndrome is associated with a riboflavin transporter defect mimicking mild MADD: a new inborn error of metabolism with potential treatment. J Inherit Metab Dis. 2011;34(1):159–164. doi:10.1007/s10545-010-9242-z. 107. Roe CR, Mochel F. Anaplerotic diet therapy in inherited metabolic disease: therapeutic potential. J Inherit Metab Dis. 2006;29:332–340. doi: 10.1007/s10545-006-0290-3.) 108. Derks TG, Reijngoud DJ, Waterham HR, Gerver WJ, Van Den Berg MP, Sauer PJ, Smit GP. The natural history of medium-chain acyl CoA dehydrogenase deficiency in the Netherlands: clinical presentation and outcome. J Pediatr. 2006;148:665–70 109. U. Spiekerkoetter &M. Lindner &R. Santer &M. Grotzke &M. R. Baumgartner &H. Boehles &A. Das &C. Haase &J. B. Hennermann &D. Karall &H. de Klerk &I. Knerr &H. G. Koch &B. Plecko &W. Ro¨schinger &K. O. Schwab &D. Scheible &F. A. Wijburg &J. Zschocke &E. Mayatepek &U. Wendel. Treatment recommendations in long-chain fatty acid oxidationdefects: consensus from a workshop. May 2009 Journal of Inherited Metabolic Disease 32(4):498-505. DOI: 10.1007/s10545-009-1126-8 110. J. Lawrence Merritt 2nd, Erin MacLeod, Agnieszka Jurecka & Bryan Hainline. Clinical manifestations and management of fatty acid oxidation disorders. Reviews in Endocrine and Metabolic Disorders volume 21, pages479–493(2020). 111. Prasun P. Multiple Acyl-CoA Dehydrogenase Deficiency. 2020 Jun 18. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2021. Available from: https://www.ncbi.nlm.nih.gov/books/NBK558236/ 112. Gillingham et al. 2006; Spiekerkoetter 2007. 113. Boer den MEJ, Wanders RJA, Morris AAM, IJLst L, Long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency: clinical presentation and follow-up of 50 patients. Pediatrics. 2002;109(1):99–104114. Boer den MEJ, Dionisi-Vici C, Mitochondrial trifunctional protein deficiency: a severe fatty acid oxidation disorder with cardiac and neurologic involvement. J Pediatr. 2003;142(6):684–689. doi:10.1067/mpd.2003.231 115. Tyni T, Johnson M, Eaton S, Pourfarzam M, Andrews R, Turnbull DM. Mitochondrial fatty acid beta-oxidation in the retinal pigment epithelium. Pediatric Research. 2002;52(4):595–600. doi:10.1203/00006450-200210000-00021 116. Oey NA, Ruiter JPN, Attié-Bitach T, IJLst L, Wanders RJA, Wijburg FA. Fatty acid oxidation in the human fetus: implications for fetal and adult disease. J Inherit Metab Dis. 2006;29(1):71–75. doi:10.1007/s10545-006-0199-x 117. Fletcher AL, Pennesi ME, harding CO, Weleber RG, Gillingham MB. Observations regarding retinopathy in mitochondrial trifunctional protein deficiencies. Molecular Genetics and Metabolism. 2012. doi:10.1016/j.ymgme.2012.02.015 118. Makhseed N., Vallance H.D., Potter M. et all. Carnitine transporter defect due to a novel mutation in the SLC22A5 gene presenting with peripheral neuropathy//J.Inherit.Metab.Dis.27(2004):778-780; https://doi.org/10.1023/B:BOLI.0000045837.23328.f4 119. Brivet M., et all. Defects in activation and transport of fatty acids .1999. https://doi.org/10.1023/A:1005552106301 120. Vianey-Saban C., Stremler N., Paut O. et al.(1995). Infantile form of carnitine palmitoyltransferase II deficiency in a girl with rapid fatal onset. J Inherit Metab Dis 18: 362–363. 121. Taroni F., Verderio E., Fiorucci S., et al. (1992). Molecular characterization of inherited carnitine palmitoyltransferase II deficiency. Proc Natl Acad Sci USA 89: 8429–8433. 122. North K.N., Hoppel C.L. et all Lethal neonatal deficiency of carnitine palmitoiltransferase II associated with dysgenesis of the brain and kidneys. J. Pediatr1995; 127:414 123. Mathur A., Sims H.F., Gopalakrishnan D. et al. Molecular heterogeneity in very long chain acyl CoA dehydrogenase deficiency causing pediatric cardiomyopathy and sudden death. Circulation 1999; 99: 1337—1343. 124. Dipti S, Childs AM, Livingston JH, Aggarwal AK, Miller M, Williams C, et al. Brown-Vialetto-Van Laere syndrome; variability in age at onset and disease progression highlighting the phenotypic overlap with Fazio-Londe disease. Brain Dev. 2005;27:443–446. 125. O"Callaghan B, Bosch AM, Houlden H. An update on the genetics, clinical presentation, and pathomechanisms of human riboflavin transporter deficiency. J Inherit Metab Dis. 2019;42:598– 607.126. Olsen R.K., Koňaříková E., Giancaspero T.A. et al. Riboflavin-responsive and -non-responsive mutations in FAD synthase cause multiple acyl-CoA dehydrogenase and combined respiratory-chain deficiency. Am J Hum Genet. 2016;98:1130–1145. 127. Schiff M., Veauville-Merllié A., Su C.H. et al. SLC25A32 mutations and riboflavin-responsive exercise intolerance. N Engl J Med. 2016;374:795–797 128. Hellebrekers D.M., Sallevelt S.C., Theunissen T.E. et al. Novel SLC25A32 mutation in a patient with a severe neuromuscular phenotype. Eur J Hum Genet. 2017;25:886–888. 129. Sklirou E., Alodaib A. N., Dobrowolski S. F. et al. Physiological Perspectives on the Use of Triheptanoin as Anaplerotic Therapy for Long Chain Fatty Acid Oxidation Disorders //Front. Genet., 15 January 2021 | https://doi.org/10.3389/fgene.2020.598760 130. Helene Ogier de Baulny, Andrea Superti-Furga. Disorders of mitochondrial fatty acid oxidation and ketone body metabolism. In: Blau N., Hoffmann G.F., Leonard J.V., Clarke J. T. R. Physician"s Guide to the Treatment and Follow-Up of Metabolic Diseases. Springer Science & Business Media, pp147-160 (http://eknygos.lsmuni.lt/springer/365/147-160.pdf) 131. А.В. Дегтярева, И.В. Никитина, И.В. Орловская, Е.Ю. Захарова, Г.В. Байдакова, О.В. Ионов, Д.Ю. Амирханова, А.В. Левадная. Дефицит ацил-коэнзим А дегидрогеназы жирных кислот с очень длинной углеродной цепью 132. Amaral, Alexandre U., Cecatto, Cristiane, Silva, Janaína C. da, Wajner, Alessandro, & Wajner, Moacir. (2017). Mechanistic Bases of Neurotoxicity Provoked by Fatty Acids Accumulating in MCAD and LCHAD Deficiencies. Journal of Inborn Errors of Metabolism and Screening, 5, e160052. Epub May 16, 2019 133. Smith E, Fernandez C, Melander O, Ottosson F. Altered Acylcarnitine Metabolism Is Associated With an Increased Risk of Atrial Fibrillation. J Am Heart Assoc. 2020 Nov 3;9(21):e016737, https://emedicine.medscape.com/article/2087425-overview#a4 134. Gillingham MB, Purnell JQ, Jordan J, Stadler D, Haqq AM, Harding CO. Effects of higher dietary protein intake on energy balance and metabolic control in children with long-chain 3-hydroxy acyl-CoA dehydrogenase (LCHAD) or trifunctional protein (TFP) deficiency. Mol Genet Metab. 2007 Jan;90(1):64-9. doi: 10.1016/j.ymgme.2006.08.002. 135. DiDonato S , Gellera C, Peluchetti D, Uziel G, Antonelli A, Lus G, Rimoldi M Normalization of short-chain acylcoenzyme A dehydrogenase after riboflavin treatment in a girl with multiple acylcoenzyme A dehydrogenase-deficient myopathy 136. Kmoch S. , Zeman J, Hrebícek M, Ryba L, Kristensen M J, Gregersen N. J Inherit Metab Dis Riboflavin-responsive epilepsy in a patient with SER209 variant form of short-chain acyl-CoA dehydrogenase . 1995;18(2):227-9. doi: 10.1007/BF00711774.137. Harding C. O. , Gillingham M. B., van Calcar S. C., Wolff J. A., Verhoeve J. N., Mills M. D. Docosahexaenoic acid and retinal function in children with long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency. 1999. May;22(3):276-80. doi: 10.1023/a:1005502626406. 138. Sun XL, Yang Z, Wang JL, Sun MN, Wu SY, Wang XY. [Correlation between severe preeclampsia and abnormal expression of long-chain fatty acid oxidative enzyme]. Zhonghua Yi Xue Za Zhi. 2011 Aug 9;91(29):2026-9. Chinese. PMID: 22093928. 139. https://www.awmf.org/uploads/tx_szleitlinien/027-006l_S3_Diagnostik-Therapie-Harnstoffzyklusstoerungen_2018-06.pdf 140. https://ojrd.biomedcentral.com/articles/10.1186/s13023-014-0130-8/tables/7 141. Дегтярева А.В., Соколова Е.В., Захарова Е.Ю., Исаева М.Х., Высоких М.Ю., Иванец Т.Ю., Дегтярев Д.Н. Гипераммониемия в практике неонатолога. Рос вестн перинатол и педиатр 2020; 65:(6): 98–107. DOI: 10.21508/1027–4065–2020–65–6–98–107 142. Knottnerus, S.J.G., Bleeker, J.C., Wüst, R.C.I. et al. Disorders of mitochondrial long-chain fatty acid oxidation and the carnitine shuttle. Rev Endocr Metab Disord 19, 93–106 (2018). https://doi.org/10.1007/s11154-018-9448-1 143. Zhu, M., Zhu, X., Qi, X., Weijiang, D., Yu, Y., Wan, H., & Hong, D. (2014). Riboflavin-responsive multiple Acyl-CoA dehydrogenation deficiency in 13 cases and a literature review in mainland Chinese patients. Journal of Human Genetics, 59(5), 256–261. doi:10.1038/jhg.2014.10 144. Wanders RJ, Visser G, Ferdinandusse S, Vaz FM, Houtkooper RH. Mitochondrial Fatty Acid Oxidation Disorders: Laboratory Diagnosis, Pathogenesis, and the Complicated Route to Treatment. J Lipid Atheroscler. 2020 Sep;9(3):313-333. https://doi.org/10.12997/jla.2020.9.3.313 145. Kompare M. & Rizzo W. B. Mitochondrial Fatty-Acid Oxidation Disorders. Seminars in Pediatric Neurology. 2008. 15(3), 140–149. doi:10.1016/j.spen.2008.05.008 146. El-Hattab, Ayman W; Li, Fang-Yuan; Shen, Joseph; Powell, Berkley R; Bawle, Erawati V; Adams, Darius J; Wahl, Erica; Kobori, Joyce A; Graham, Brett; Scaglia, Fernando; Wong, Lee-Jun (2010). Maternal systemic primary carnitine deficiency uncovered by newborn screening: Clinical, biochemical, and molecular aspects. Genetics in Medicine, 12(1), 19–24. doi:10.1097/GIM.0b013e3181c5e6f7 147. Lisa A. Schimmenti; Eric A. Crombez; Bernd C. Schwahn; Bryce A. Heese; Timothy C. Wood; Richard J. Schroer; Kristi Bentler; Stephen Cederbaum; Kiki Sarafoglou; Mark McCann; Piero Rinaldo; Dietrich Matern; Cristina Amat di San Filippo; Marzia Pasquali; Susan A. Berry; Nicola Longo (2007). Expanded newborn screening identifies maternal primary carnitine deficiency. , 90(4), 0–445. doi:10.1016/j.ymgme.2006.10.003148. S. E. Olpin, J. Allen, J. R. Bonham et al. Features of carnitine palmitoyltransferase type I de¢ciency J. Inherit. Metab. Dis. 24 (2001) 35^42 149. Nyhan, W.L., Hoffmann, G.F., Al-Aqeel, A.I., & Barshop, B.A. (2019). Atlas of Inherited Metabolic Diseases (4th ed.). CRC Press. https://doi.org/10.1201/9781315114033 150. F Feillet , G Steinmann, C Vianey-Saban, C de Chillou, N Sadoul, E Lefebvre, M Vidailhet, P E Bollaert Adult presentation of MCAD deficiency revealed by coma and severe arrythmias Intensive Care Med (2003) 29:1594–1597 DOI 10.1007/s00134-003-1871-3 151. https://www.newenglandconsortium.org/mcadd 152. Savy N, Brossier D, Brunel-Guitton C, Ducharme-Crevier L, Du Pont-Thibodeau G, Jouvet P. Acute pediatric hyperammonemia: current diagnosis and management strategies. Hepat Med. 2018 Sep 12;10:105-115. doi: 10.2147/HMER.S140711. PMID: 30254497; PMCID: PMC6140721. 153. https://www.uptodate.com/contents/insulin-regular-pediatric-drug-information? search=insulin&topicRef=85908&source=see_link 154. Matoori S, Leroux JC. Recent advances in the treatment of hyperammonemia. Adv Drug Deliv Rev. 2015 Aug 1;90:55-68. 155. Magoulas PL, El-Hattab AW. Systemic primary carnitine deficiency: an overview of clinical manifestations, diagnosis, and management. Orphanet J Rare Dis. 2012;7:68. 156. El-Hattab AW. Systemic Primary Carnitine Deficiency. In: GeneReviews®. University of Washington, Seattle, Seattle (WA); 1993. PMID: 22420015. 157. Gillingham MB, Weleber RG, Neuringer M, et al. Effect of optimal dietary therapy upon visual function in children with long-chain 3-hydroxyacyl CoA dehydrogenase and trifunctional protein deficiency. Mol Genet Metab. 2005 Sep-Oct;86(1-2):124-33. 158. Gataullina S, Delonlay P, Lemaire E, et al. Seizures and epilepsy in hypoglycaemia caused by inborn errors of metabolism. Dev Med Child Neurol. 2015 Feb;57(2):194-9. 159. Неонатальный скрининг: национальное руководство / под ред. С.И. Куцева. Москва : ГЭОТАР-Медиа, 2023. — 360 с. — (Серия «Национальные руководства»). — DOI: 10.33029/9704-7737-3-NEO-2023-1-360. 160. Umpierrez GE, Hellman R, Korytkowski MT, Kosiborod M, Maynard GA, Montori VM, Seley JJ, Van den Berghe G; Endocrine Society. Management of hyperglycemia in hospitalized patients in non-critical care setting: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2012 Jan;97(1):16-38. doi: 10.1210/jc.2011-2098
Информация
Список сокращений
Термины и определения
Катаболизм – процесс распада сложных веществ;
Рабдомиолиз – разрушение клеток мышечной ткани;
Синдром Рейе – заболевание, проявляющееся острой невоспалительной (токсической) энцефалопатией в сочетании с жировой дистрофией внутренних органов, преимущественно печени (острый микровезикулярный стеатоз);
HELLP-синдром – тяжелое осложнение беременности, для которого характерна триада признаков: гемолиз, повреждение печеночной паренхимы с повышением активности печеночных трансаминаз и тромбоцитопения.
Прогноз состояния и уровня психического развития пациентов зависит от тяжести заболевания, степени поражения внутренних органов (сердце, печень) и ЦНС, сроков начала лечения и эффективности интенсивной терапии при метаболической декомпенсации. Рано манифестирующая системная форма заболевания обычно имеет более тяжелое течение и менее благоприятный прогноз.
| № п/п | Критерии оценки качества | Оценка выполнения |
|---|---|---|
| 1. | Выполнено комплексное определение концентрации на аминокислоты и ацилкарнитины в крови методом тандемной масс-спектрометрии (при установлении диагноза) | Да/Нет |
| 2. | Выполнено молекулярно-генетическое исследование мутаций в генах, ответственных за β-окисление жирных кислот, метаболизм и транспорт карнитина (при установлении диагноза пациентам с биохимическими признаками нарушения митохондриального β-окисления жирных кислот) | Да/Нет |
| 3. | Выполнен прием (осмотр, консультация) врача-генетика первичный (при установлении диагноза) | Да/Нет |
| 4. | Выполнено назначение сухой специализированной смеси с повышенным содержанием СЦТ (при нарушении окисления жирных кислот с длинной и очень длинной цепью) | Да/Нет |
Критерии оценки качества специализированной медицинской помощи
| № п/п | Критерии оценки качества | Оценка выполнения |
|---|---|---|
| 1. | Выполнено комплексное определение концентрации на аминокислоты и ацилкарнитины в крови методом тандемной масс-спектрометрии (при установлении диагноза) | Да/Нет |
| 2. | Выполнено назначение сухой специализированной смеси с повышенным содержанием СЦТ (при нарушении окисления жирных кислот с длинной и очень длинной цепью) | Да/Нет |
Приложение А1. Состав рабочей группы по разработке и пересмотру клинических рекомендаций
- Анисимова Инга Вадимовна — к.м.н., заведующая отделом организации медицинской помощи, врач-генетик ФГБНУ «Медико-генетический научный центр им. академика Н.П. Бочкова», член Ассоциации медицинских генетиков
- Багаева Мадлена Энверовна — к.м.н., с.н.с. отделения педиатрической гастроэнтерологии, гепатологии и диетотерапии ФГБУН "ФИЦ питания и биотехнологии", ассистент кафедры ФГАОУ ВО РНИМУ им. Н.И.Пирогова Минздрава России.
- Байдакова Галина Викторовна — к.б.н., заведующая Центром коллективного пользования "Метаболом", в.н.с. лаборатории наследственных болезней обмена ФГБНУ "Медико-генетический научный центр им. академика Н.П.Бочкова", член Российского общества медицинских генетиков.
- Баранов Александр Александрович — академик РАН, профессор, д.м.н.; почетный президент Союза педиатров России, советник руководителя НИИ педиатрии и охраны здоровья детей ЦКБ РАН, профессор кафедры педиатрии и детской ревматологии ФГАОУ «Первый МГМУ им. И.М. Сеченова» Минздрава России (Сеченовский Университет), главный внештатный специалист педиатр Минздрава России.
- Бушуева Татьяна Владимировна — д.м.н., ФГАУ "Научный медицинский исследовательский центр здоровья детей" МЗ РФ, член ассоциации медицинских генетиков, член ПНО Ассоциация здоровье детей.
- Вашакмадзе Нато Джумберовна — д.м.н., руководитель отдела орфанных болезней и профилактики инвалидизирующих заболеваний НИИ педиатрии и охраны здоровья детей НКЦ №2 ФГБНУ «РНЦХ им акад. Б. В. Петровского», профессор кафедры факультетской педиатрии педиатрического факультета ФГБОУ ВО «РНИМУ им. Н.И. Пирогова» Минздрава России
- Вишнёва Елена Александровна — д.м.н., заместитель руководителя НИИ педиатрии и охраны здоровья детей ЦКБ РАН Минобрнауки по научной работе, доцент кафедры факультетской педиатрии педиатрического факультета ФГБОУ ВО «РНИМУ им. Н.И. Пирогова» Минздрава России, член Союза педиатров России.
- Дегтярева Анна Владимировна — д.м.н., профессор, заведующая отделом педиатрии, ФГБУ «НМИЦ акушерства, гинекологии и перинатологии им. академика В.И. Кулакова», член Российского общества неонатологов
- Докшукина Алина Алексеевна — врач-неонатолог, младший научный сотрудник отдела педиатрии ФГБУ «НМИЦ акушерства, гинекологии и перинатологии им. академика В.И. Кулакова» член Российского общества неонатологов
- Зарубина Вера Владимировна —врач-генетик Морозовской ДГКБ ДЗМ
- Журкова Наталия Вячеславовна — к.м.н., ведущий научный сотрудник НИИ педиатрии и охраны здоровья детей НКЦ №2 ФГБНУ «РНЦХ им акад. Б. В. Петровского», член Союза педиатров России, член Ассоциации медицинских генетиков
- Захарова Екатерина Юрьевна — д.м.н., заведующая лабораторией наследственных болезней обмена ФГБНУ "Медико-генетический научный центр им. академика Н.П. Бочкова", член Российского общества медицинских генетиков, член европейского общества по изучению наследственных болезней обмена веществ (SSIEM).
- Кузенкова Людмила Михайловна — д.м.н., заведующая отделением психоневрологии и психосоматической патологии ФГАУ "Научный медицинский исследовательский центр здоровья детей" МЗ РФ, член Союза педиатров России
- Куцев Сергей Иванович — академик РАН, д.м.н., директор ФГБНУ "Медико-генетический научный центр им. академика Н.П.Бочкова", Президент Ассоциации медицинских генетиков
- Лаврова Алла Евгеньевна — д.м.н., директор института педиатрии, заведующий педиатрическим отделением N 2, Университетская клиника ФГБОУ ВО «Приволжский исследовательский медицинский университет» МЗ РФ, отличник здравоохранения.
- Михайлова Светлана Витальевна — д.м.н., заведующая отделением ФГБУ «Российская Детская Клиническая Больница» МЗ РФ
- Намазова-Баранова Лейла Сеймуровна — акад. РАН, профессор, д.м.н., президент Союза педиатров России; паст-президент EPA/UNEPSA; руководитель НИИ педиатрии и охраны здоровья детей ЦКБ РАН, заведующая кафедрой факультетской педиатрии педиатрического факультета ФГБОУ ВО «РНИМУ им. Н.И. Пирогова» Минздрава России, главный внештатный детский специалист по профилактической медицине Минздрава России
- Назаренко Людмила Павловна — профессор, д.м.н., заместитель директора по научной и лечебной работе, руководитель лаборатории наследственной патологии НИИ медицинской генетики, Томского НИМЦ РАН, член Ассоциации медицинских генетиков
- Николаева Екатерина Александровна — д.м.н., главный научный сотрудник отдела клинической генетики Научно-исследовательского клинического института педиатрии имени акад. Ю.Е. Вельтищева, проф. кафедры инновационной педиатрии и детской хирургии Института непрерывного образования и профессионального развития ФГАОУ ВО РНИМУ им. Н.И. Пирогова Минздрава России
- Первунина Татьяна Михайловна — д.м.н., врач-педиатр, врач-детский кардиолог, доцент кафедры педиатрии медицинского факультета СПБГУ, директор института педиатрии и перинатрлогии ФГБУ " НМИЦ им. В.А. Алмазова.
- Печатникова Наталья Леонидовна — руководитель Городского Центра орфанных и других редких заболеваний у детей и подростков ГБУЗ «Морозовская ДГКБ ДЗМ».
- Репина Светлана Афанасьевна — к.м.н., врач-генетик ФГБНУ "Медико-генетический научный центр им. академика Н.П.Бочкова ", член Российского общества медицинских генетиков, член Ассоциации медицинских генетиков
- Свиридова Валерия Валерьевна — врач-генетик отдела организации медицинской помощи ФГБНУ «Медико-генетический научный центр им. академика Н.П. Бочкова», м.н.с лаборатории мутагенеза ФГБНУ «Медико-генетический научный центр им. академика Н.П. Бочкова»
- Сеитова Гульнара Наримановна — к.м.н., главный врач Медико-генетического центра (Генетической клиники) НИИ медицинской генетики, Томского НИМЦ РАН
- Селимзянова Лилия Робертовна — к.м.н., заведующая отделом НИИ педиатрии и охраны здоровья детей НКЦ №2 ФГБНУ «РНЦХ им акад. Б. В. Петровского», доцент кафедры педиатрии и детской ревматологии ФГАОУ «Первый МГМУ им. И. М. Сеченова» Минздрава России (Сеченовский Университет), доцент кафедры факультетской педиатрии педиатрического факультета ФГБОУ ВО «РНИМУ им. Н. И. Пирогова» Минздрава России, член Союза педиатров России
- Смирнова Ольга Яковлевна – врач-генетик, старший научный сотрудник отдела стандартизации и изучения основ доказательной медицины НИИ педиатрии и охраны здоровья детей ЦКБ РАН
- Строкова Татьяна Викторовна — д.м.н., профессор РАН, заведующая отделением педиатрической гастроэнтерологии, гепатологии и диетотерапии ФГБУН "ФИЦ питания и биотехнологии",", заведующая кафедрой «Гастроэнтерологии и диетологии» ФГАОУ ВО РНИМУ им. Н.И.Пирогова Минздрава России.
- Субботин Дмитрий Михайлович — врач-генетик ФГБНУ «Медико-генетический научный центр им. академика Н. П. Бочкова», член Ассоциации медицинских генетиков
- Таран Наталия Николаевна — к.м.н., старший научный сотрудник отделения педиатрической гастроэнтерологии, гепатологии и диетотерапии ФГБУН "ФИЦ питания и биотехнологии"", ассистент кафедры ФГАОУ ВО РНИМУ им. Н.И.Пирогова Минздрава России.
- Фисенко Андрей Петрович – д.м.н., профессор, Заслуженный врач Российской Федерации, директор ФГАУ "Национальный медицинский исследовательский центр здоровья детей" Минздрава России.
Представители общественных пациентских организаций:
Авторы подтверждают отсутствие финансовой поддержки/конфликта интересов, который необходимо обнародовать.
Приложение А2. Методология разработки клинических рекомендаций
Целевая аудитория данных клинических рекомендаций:
- Врачи общей практики (семейные врачи);
- Врачи- педиатры;
- Врачи-терапевты;
- Врачи-генетики;
- Врачи-лабораторные генетики;
- Врачи-кардиологи;
- Врачи-детские кардиологи;
- Врачи- неврологи;
- Врачи функциональной диагностики;
- Медицинские психологи;
- Студенты медицинских ВУЗов;
- Обучающиеся в ординатуре и интернатуре.
Таблица 1. Шкала оценки уровней достоверности доказательств (УДД) для методов диагностики (диагностических вмешательств)
| УДД | Расшифровка |
|---|---|
| 1 | Систематические обзоры исследований с контролем референсным методом или систематический обзор рандомизированных клинических исследований с применением мета-анализа |
| 2 | Отдельные исследования с контролем референсным методом или отдельные рандомизированные клинические исследования и систематические обзоры исследований любого дизайна, за исключением рандомизированных клинических исследований, с применением мета-анализа |
| 3 | Исследования без последовательного контроля референсным методом или исследования с референсным методом, не являющимся независимым от исследуемого метода или нерандомизированные сравнительные исследования, в том числе когортные исследования |
| 4 | Несравнительные исследования, описание клинического случая |
| 5 | Имеется лишь обоснование механизма действия или мнение экспертов |
Таблица 2. Шкала оценки уровней достоверности доказательств (УДД) для методов профилактики, лечения и реабилитации (профилактических, лечебных, реабилитационных вмешательств)
| УДД | Расшифровка |
|---|---|
| 1 | Систематический обзор РКИ с применением мета-анализа |
| 2 | Отдельные РКИ и систематические обзоры исследований любого дизайна, за исключением РКИ, с применением мета-анализа |
| 3 | Нерандомизированные сравнительные исследования, в т.ч. когортные исследования |
| 4 | Несравнительные исследования, описание клинического случая или серии случаев, исследования «случай-контроль» |
| 5 | Имеется лишь обоснование механизма действия вмешательства (доклинические исследования) или мнение экспертов |
Таблица 3. Шкала оценки уровней убедительности рекомендаций (УУР) для методов профилактики, диагностики, лечения и реабилитации (профилактических, диагностических, лечебных, реабилитационных вмешательств)
| УУР | Расшифровка |
|---|---|
| A | Сильная рекомендация (все рассматриваемые критерии эффективности (исходы) являются важными, все исследования имеют высокое или удовлетворительное методологическое качество, их выводы по интересующим исходам являются согласованными) |
| B | Условная рекомендация (не все рассматриваемые критерии эффективности (исходы) являются важными, не все исследования имеют высокое или удовлетворительное методологическое качество и/или их выводы по интересующим исходам не являются согласованными) |
| C | Слабая рекомендация (отсутствие доказательств надлежащего качества (все рассматриваемые критерии эффективности (исходы) являются неважными, все исследования имеют низкое методологическое качество и их выводы по интересующим исходам не являются согласованными) |
Порядок обновления клинических рекомендаций.
Приложение А3. Справочные материалы, включая соответствие показаний к применению и противопоказаний, способов применения и доз лекарственных препаратов, инструкции по применению лекарственного препарата
- Приказ Минздрава России от 21 апреля 2022 года № 274н «Об утверждении Порядка оказания медицинской помощи пациентам с врожденными и (или) наследственными заболеваниями»
- Федеральный закон "Об основах охраны здоровья граждан в Российской Федерации" (N 323-ФЗ от 21.11.2011).
- Постановление Правительства РФ от 30 июля 1994 г. N 890 "О государственной поддержке развития медицинской промышленности и улучшении обеспечения населения и учреждений здравоохранения лекарственными средствами и изделиями медицинского назначения" (с изменениями и дополнениями)
- Приказ Министерства здравоохранения Российской Федерации от 15 декабря 2014 г. N 834н "Об утверждении унифицированных форм медицинской документации, используемых в медицинских организациях, оказывающих медицинскую помощь в амбулаторных условиях, и порядков по их заполнению".
- Приказ Минздрава России от 12 ноября 2021 г. № 1051н «Об утверждении порядка дачи информированного добровольного согласия на медицинское вмешательство и отказа от медицинского вмешательства, формы информированного добровольного согласия на медицинское вмешательство и формы отказа от медицинского вмешательства»
- Приказ Минздрава России от 24 ноября 2021 г. № 1094н "Об утверждении порядка назначения лекарственных препаратов, форм рецептурных бланков на лекарственные препараты, порядка оформления указанных бланков, их учета и хранения, форм бланков рецептов, содержащих назначение наркотических средств или психотропных веществ, порядка их изготовления, распределения, регистрации, учета и хранения, а также правил оформления бланков рецептов, в том числе в форме электронных документов"
- Приказ Министерства здравоохранения Российской Федерации от 16 мая 2019 г. N 302н "Об утверждении Порядка прохождения несовершеннолетними диспансерного наблюдения, в том числе в период обучения и воспитания в образовательных организациях "
- Распоряжение Правительства РФ от 31 декабря 2018 г. № 3053-р "Об утверждении перечней медицинских изделий, имплантируемых в организм человека при оказании медицинской помощи в рамках программы государственных гарантий бесплатного оказания гражданам медицинской помощи и отпускаемых по рецептам на медицинские изделия при предоставлении набора социальных услуг"
Информация о лекарственных средствах: https://grls.rosminzdrav.ru/
Основные нормативно-правовые акты, регулирующие оказание паллиативной медицинской помощи
- Федеральный закон "О внесении изменений в Федеральный закон "Об основах охраны здоровья граждан в Российской Федерации" по вопросам оказания паллиативной медицинской помощи" от 06.03.2019 № 18-ФЗ.
- Приказ Минздрава России № 345н, Минтруда России от 31.05.2019 № 372н «Об утверждении положения об организации оказания паллиативной медицинской помощи, включая порядок взаимодействия медицинских организаций, организаций социального обслуживания и общественных объединений, иных некоммерческих организаций, осуществляющих свою деятельность в сфере охраны здоровья».
- Приказ Минздрава России № 348н от 31 мая 2019 года «Об утверждении перечня медицинских изделий, предназначенных для поддержания органов и систем организма человека, предоставляемых для использования на дому».
- Приказ Минздрава России № 505н от 10 июля 2019 года «Об утверждении Порядка передачи от медицинской организации пациенту (его законному представителю) медицинских изделий, предназначенных для поддержания функций органов и систем организма человека, для использования на дому при оказании паллиативной медицинской помощи».
-
Приказ МЗ РФ № 831 от 3 октября 2019 года «Об утверждении ведомственной целевой программы «Развитие системы оказания паллиативной медицинской помощи».Прочие информационные ресурсы и нормативно-правовые документы, с учетом которых разработаны клинические рекомендации:
- Международная классификация болезней, травм и состояний, влияющих на здоровье (МКБ – 10);
- Приказ МЗ РФ от 2 мая 2023 г. № 205н «Об утверждении номенклатуры должностей медицинских работников и фармацевтических работников»
- Приказ МЗ РФ от 23 июля 2010 г. № 541н. Единый квалификационный справочник должностей руководителей, специалистов и служащих, раздел Квалификационные характеристики должностей работников в сфере здравоохранения.
- Федеральный закон от 25.12.2018 № 489 489-ФЗ «О внесении изменений в статью 40 Федерального закона "Об обязательном медицинском страховании в Российской Федерации" и Федеральный закон "Об основах охраны здоровья граждан в Российской Федерации" по вопросам клинических рекомендаций».
- Приказ Минздрава России № 103н от 28.02.2019 г. «Об утверждении порядка и сроков разработки клинических рекомендаций, их пересмотра, типовой формы клинических рекомендаций и требований к их структуре, составу и научной обоснованности, включаемой в клинические рекомендации информации».
- Приказ Минздрава России от 13.10.2017 N 804н "Об утверждении номенклатуры медицинских услуг".
- Приказ Министерства труда и социальной защиты РФ от 27 августа 2019 г. n 585н "О классификациях и критериях, используемых при осуществлении медико-социальной экспертизы граждан федеральными государственными учреждениями медико-социальной экспертизы";
- Приказ Министерства здравоохранения и социального развития Российской Федерации «О порядке применения лекарственных средств у больных по жизненным показаниям» от 9 августа 2005 г. № 494
- Информационное письмо Минздрава России по возможности закупки лекарственного препарата по торговому наименованию (https://www.rosminzdrav.ru/news/2019/12/18/13043-minzdrav-podgotovil-informatsionnoe-pismo-po-vozmozhnosti-zakupki-lekarstvennogo-preparata-po-torgovomu-naimenovaniyu);
- Распоряжение Правительства РФ от 11.12.2023 N 3551-р «Об утверждении перечня специализированных продуктов лечебного питания для детей-инвалидов»
Приложение А4. Классификация наследственных нарушений окисления жирных кислот
| Заболевание | Частота | Ген | Локус | OMIM |
|---|---|---|---|---|
| Нарушения транспорта жирных кислот | ||||
| Первичный дефицит карнитина (OCTN2) | 1:22000-1:7700 | SLC22A5 | 5q31.1 | 212140 |
| Карнитин пальмитоилтрансферазы 1 дефицит (СРТ1) | редко |
CPT1A
|
11q13.1 | 600528 |
| Карнитин-ацилкарнитин транслоказы дефицит (CACT) | редко | SLC25A20 | 3p21.31 | 212138 |
| Карнитин пальмитоилтрансферазы 2 дефицит (СРТ2) | редко | СРТ2 | 1p32 | 600650 |
| Нарушение ß-окисления жирных кислот | ||||
| Короткоцепочечной ацил-КоА дегидрогеназы дефицит (SCAD) | 1:25000-1:220000 | ACADS | 12q22-qter | 201470 |
| Среднецепочечной ацил-КоА дегидрогеназы дефицит (MCAD) | 1:15000 | ACADM | 1p31 | 201450 |
| Очень длинноцепочечной ацил-КоА дегидрогеназы дефицит (VLCAD) | 1:85000 | ACADVL | 17p13 | 201475 |
| Короткоцепочечной 3-гидроксиацил- КоА дегидрогеназы дефицит (SCHAD) | редко |
SCHAD
|
4q22-26 | 601609 |
| Длинноцепочечной 3-гидроксиацил- КоА дегидрогеназы дефицит (LCHAD) | 1:250000-1:750000 |
HADHA
|
2p23 | 609016 |
| Митохондриального трифункционального белка дефицит (TFP) | 1:250000-1:750000 | HADHA, HADHВ | 2p23 | 609015 |
| Множественный дефицит ацил-КоА дегидрогеназ дефицит//глутаровая ацидурия тип 2 (ГА2) | редко |
ETF-B
ETF-A
ETF-DH
|
19q13.3, 15q23-q25,
4q32-qter
|
231690, 231675 |
Приложение А5. Клинические проявления нарушений окисления жирных кислот
| Клинические признаки | Нарушения |
|---|---|
| Энцефалопатия (судороги, летаргия, кома) | Все формы, кроме мышечного типа недостаточности карнитина |
| Поражение печени | Все формы |
| Мышечная слабость, гипотония | Все формы |
| Миалгия, рабдомиолиз, изменение цвета мочи (красновато-бурая) | CPT 2, CAСT, VLCAD, TFP, LCHAD, ГА2 |
| Кардиомиопатия | Карнитина недостаточность, CPT 1 (редко), CPT 2, CAСT, VLCAD, TFP, LCHAD, ГА2 |
| Нарушения ритма сердца | CPT 2, CAСT, VLCAD, TFP, LCHAD, MCAD, ГА2 |
| Синдром внезапной детской смерти | Карнитина недостаточность, CPT 2, CAСT, VLCAD, TFP, LCHAD, MCAD, ГА2 |
| Лицевые дизморфии | CPT 2, ГА2 |
| Тубулярные нарушения | CPT 1, CPT 2, LCHAD, ГА2 |
| Почечная недостаточность | CPT 1, CPT 2, CAСT, VLCAD, TFP, LCHAD, ГА2 |
| Наличие кист в почках | CPT 2, ГА2 |
| Увеличение почек | CPT 1, CPT 2, ГА2 |
| Боли в животе, диарея | Карнитина недостаточность, CPT 1, TFP, LCHAD |
| Периферическая нейропатия | TFP, LCHAD |
| Пигментный ретинит, помутнение хрусталика (редко) | TFP, LCHAD |
| Необычный запах мочи (типа «потных ног») | ГА2 |
| Осложнения беременности у матерей (неукротимая рвота, увеличение печени, повышение активности трансаминаз, гипербилирубинемия, жировая дистрофия печени) | CPT 1, TFP, LCHAD |
Приложение А5а.Общие клинические проявления FAOD
| Клинические симптомы | CPT1 | OCTN2 | CACT | CPT2 | VLCAD | TFP | LCHAD |
|---|---|---|---|---|---|---|---|
| Летальность | Высокая | + | 37-43% | 76% | 38% | ||
|
Гипогликемии/
Вялость, сонливость
|
+ | + | + | + | 30-43% | 57% | 30% |
| Кардиомиопатии/ дыхательные расстройства | - | + | + | + | 52-36% | 73% | 49% |
| Нарушение сердечного ритма | - | - | + | 18% | 10% | 18% | |
| Миопатия/крампи/рабдомиолиз | - | + | 96% | 20-4% | 83% | ||
| Мышечная гипотония | - | + | - | - | - | 100-80% | 61% |
| Гепатомегалия/гепатопатия | + | + | + | + | 61-96% | 60% | 78% |
| Полиневропатия | - | - | - | - | - | 70% | 3% |
| Ретинопатия | - | - | - | - | - | 11% | 29% |
Приложение А6. Лабораторные изменения при нарушениях окисления жирных кислот
| Заболевание | Лабораторные изменения |
|---|---|
| Первичный дефицит карнитина |
Кровь: гипокетотическая гипогликемия на фоне голода, ↑ КФК, ↑АСТ, ↑АЛТ
Моча: Дикарбоновые кислоты в норме или несколько повышены
Ацилкарнитины: ↓: ↓С0<10, суммарные ацилкарнитины <5
|
| Карнитин пальмитоилтрансферазы 1А дефицит (СРТ1) |
Кровь: гипокетотическая гипогликемия на фоне голода, ↑ КФК, ↑АСТ, ↑АЛТ ↑↑NH3;
Коагулопатия;
Моча: отсутствие повышения дикарбоновых кислот
Ацилкарнитины: ↑С0, С0/(С16+С18)
↓С16, С18, С18:1
|
| Карнитин-ацилкарнитин транслоказы дефицит (CACT) |
Кровь: гипокетотическая гипогликемия на фоне голода, ↑ КФК, ↑АСТ, ↑АЛТ, ↑лактат, ↑ NH3
Коагулопатия
Моча: может отмечаться повышение дикарбоновых кислот
Ацилкарнитины: ↑ С16, С16:1, С18, С18:1
↓С0
|
| Карнитин пальмитоилтрансферазы 2 дефицит (СРТ2) |
Кровь: гипокетотическая гипогликемия на фоне голода ↑ КФК, ↑АСТ, ↑АЛТ
Моча: миоглобинурия могут повышаться дикарбоновые кислоты
Ацилкарнитины: ↑ С16, С16:1, С18, С18:1
↓С0
|
| Короткоцепочечной ацил-КоА дегидрогеназы дефицит (SCAD) |
Кровь: метаболический ацидоз, гипогликемия
Моча: этилмалоновая кислота
Ацилкарнитины: ↑ С4
|
| Среднецепочечной ацил-КоА дегидрогеназы дефицит (MCAD) |
Кровь: гипокетотическая гипогликемия на фоне голода, ↑АСТ, ↑АЛТАЛТ
Моча: С6-С10 дикарбоновые кислоты, суберилглицин, гексаноилглицин
Ацилкарнитины: ↑С8, С10, C10:1
↓С0 (или норма)
|
| Очень длинноцепочечной ацил-КоА дегидрогеназы дефицит (VLCAD) |
Кровь: гипокетотическая гипогликемия на фоне голода, ↑ КФК, ↑АСТ, ↑АЛТ, ↑ NH3
Моча: С6-С14 дикарбоновые кислоты
Ацилкарнитины: ↑С12:1, С14, С14:1, С16, С16:1
|
| Короткоцепочечной 3-гидроксиацил- КоА дегидрогеназы дефицит (SCHAD) |
Кровь: гипокетотическая гипогликемия на фоне голода, ↑инсулин, ↑NH3
Моча: 3-гидроксиглутаровая кислота
Ацилкарнитины: ↑ С4ОН
|
| Длинноцепочечной 3-гидроксиацил- КоА дегидрогеназы дефицит (LCHAD) |
Кровь: гипокетотическая гипогликемия на фоне голода, ↑ КФК, ↑АСТ, ↑АЛТ, ↑ NH3
Моча: С6-С14 дикарбоновые кислоты;
Ацилкарнитины:↑:↑С16:1-ОН, С16-ОН, С18:1-ОН, С18OH
|
| Митохондриального трифункционального белка дефицит (TFP) |
Кровь: гипокетотическая гипогликемия на фоне голода, ↑ КФК, ↑АСТ, ↑АЛТ, ↑ NH3
Моча: С6-С14 дикарбоновые кислоты;
Ацилкарнитины:↑:↑С16:1-ОН, С16-ОН, С18:1-ОН, С18OH
|
| Множественный дефицит ацил-КоА дегидрогеназ дефицит глутаровая ацидурия тип II (ГА2ГА2ГА2ГА2) |
Сыворотка: гипокетотическая гипогликемия на фоне голода,гипокетотическая гипогликемия на фоне голода, ↑ КФК, ↑АСТ, ↑АЛТ, ↑ NH3, метаболический ацидоз
Моча: ↑С5-С10 дикарбоновые кислоты, молочная, глутаровая, этилмолоновая кислоты.
Ацилкарнитины: ↑С4-С18
|
Приложение А7. Забор биоматериала для диагностики в пятнах крови

Алгоритм действий медицинского персонала при взятии образцов крови
- вымыть руки (гигиенический уровень), надеть перчатки;
- вымыть руки пациента (пятку ребенка, в случае, если кровь берется из пятки);
- протереть область прокалывания стерильной салфеткой, смоченной 70% спиртом, промокнуть сухой стерильной салфеткой; - проколоть стерильным одноразовым скарификатором;
- снять первую каплю крови стерильным сухим тампоном из нетканного материала или марли, или салфеткой марлевой медицинской стерильной;
- мягко надавить для получения второй капли крови;
- приложить перпендикулярно тест-бланк к капле крови и пропитать его кровью насквозь;
- аналогичным образом нанести на тест-бланк 6-8 капель, вид пятен крови должен быть одинаковым с обеих сторон.
- высушить тест-бланк в горизонтальном положении на чистой обезжиренной поверхности не менее 4 ч без применения тепловой обработки и попадания прямых солнечных лучей;
- упаковать тест-бланки в чистый конверт таким образом, чтобы пятна крови не соприкасались.
Особенности при инфузионной терапии
Не допускается забор крови
- сразу после проведения пациенту инфузионной терапии;
- сразу после заменного переливания крови.
Хранение и транспортировка биоматериала
Образцы высушенных пятен крови можно хранить в обычной камере холодильника при +40С до отправки. Срок хранения до момента отправки не должен превышать 7 дней. Если хранить дольше и при более высокой температуре, то это приведет к ложноположительным результатам.
Приложение А8. Максимальная продолжительность голодания (при стабильных метаболических состояниях)
Макисмальные интервалы между кормлениями для пациентов с нарушениями окисления длинноцепочечных жирных кислот [66].
| возраст | интервалы между кормлениями |
|---|---|
| Новорожденные | 3 часа днем и ночью |
| < 6 месяцев | 4 часа днем и ночью |
| > 6 месяцев-12 месяцев | 4 часа днем, и 6-8 часов ночью |
| > 12 месяцев | 4 часа днем, 10 часов ночью |
Внимание! Если у детей нет проблем со вксармливанием, ночное голодание может быть продлено до 6 часов, начиная с 3-месячного возраста и до 8 часов, начиная с 6-месячного возраста. С осторожностью разрешать голодание более 10 часов даже после первого года лечения. Риск ночной гипогликемии снижается по мере взросления ребенка, и большинство детей после 4 лет, могут переносить периоды без приема пищи 10-12 часов если чувствуют себя хорошо
Макисмальные интервалы между кормлениями для пациентов с дефицитом длинноцепочечной 3-ОН ацилКоА дегидрогеназы жирных кислот LCHAD (https://www.ncbi.nlm.nih.gov/books/NBK583531/)
| возраст | рекомендации |
|---|---|
| Новорожденные – 3 мес | частое кормление (каждые 2-3 часа), так же ночью |
| 4–12 мес | Каждые 3часа и можно увеличить до 4 часов, если переносимость сохраняется в течение 6 мес. |
| > 6 месяцев-12 месяцев | дневное кормление каждые 4 часа; Ночное голодание может быть постепенно увеличено до 6-8 часов к 12 мес. |
| 1–3 года | интервал дневного кормления 4 часа; можно попробовать ночное голодание до 10 часов. |
| Старше 3 лет |
можно попробовать ночное голодание продолжительностью до 12 часов.
|
Макисмальные интервалы между кормлениями для пациентов с дефицитом среднецепочечной дегидрогеназы жирных кислот (https://www.ncbi.nlm.nih.gov/books/NBK560837/)
| Возраст | Безопасный период голодания |
|---|---|
| При положительных результатах скрининга в возрасте от 0 до 4 месяцев* | 4 часа |
| От 5 месяцев до 12 | Постепенно возрастает от 5 до 12 часов (каждый месяц добавляется 1 час) |
| От 12 месяцев и старше | 12 часов |
Внимание! Рекомендуют проверять уровень глюкозы в крови перед кормлением утром, чтобы определить переносимость ночного голодания и при необходимости составить индивидуальные рекомендации.
Приложение А9. Среднесуточные нормы физиологических потребностей в энергии
|
Среднесуточные нормы физиологических потребностей в энергии для детей
согласно FAO/WHO/UNU 2001 (WHO Technical Report Series 17-24okt 2001)
|
||
|---|---|---|
| Возраст (мес.) | Энергия (ккал/кг) муж. | Энергия (ккал/кг) жен |
| 0-1 | 113 | 107 |
| 1-2 | 104 | 101 |
| 2-3 | 95 | 94 |
| 3-4 | 82 | 84 |
| 4-5 | 81 | 83 |
| 5-6 | 81 | 82 |
| 6-7 | 79 | 78 |
| 7-8 | 79 | 78 |
| 8-9 | 79 | 78 |
| 9-10 | 80 | 79 |
| 10-11 | 80 | 79 |
| 11-12 | 81 | 79 |
| Возраст (г.) | ||
| 1-2 | 82,4 | 80,1 |
| 2-3 | 83,6 | 80,6 |
| 3-4 | 79,7 | 76,5 |
| 4-5 | 76,8 | 73,9 |
| 5-6 | 74,5 | 71,5 |
| 6-7 | 72,6 | 69,3 |
| 7-8 | 70,5 | 66,7 |
| 8-9 | 68,5 | 63,8 |
| 9-10 | 66,6 | 60,8 |
| 10-11 | 64,6 | 57,8 |
| 11-12 | 62,4 | 54,8 |
| 12-13 | 60,2 | 52,0 |
| 13-14 | 57,9 | 49,3 |
| 14-15 | 55,6 | 47,0 |
| 15-16 | 53,4 | 45,3 |
| 16-17 | 51,6 | 44,4 |
| 17-18 | 50,3 | 44,1 |
| Среднесуточные нормы физиологических потребностей в энергии для людей разных возрастов согласно FAO/WHO/UNU 2007 (WHO Technical Report Series 2007) | ||
|---|---|---|
| Возраст (г.) | Энергия (ккал/кг) муж. | Энергия (ккал/кг) жен |
| Взрослые, ведущие образ жизни средней активности, вес 70кг | ||
| 18-29 | 43,7 | 38,0 |
| 30-59 | 41,8 | 35,3 |
| Взрослые, ведущие образ жизни средней активности, вес 50кг | ||
| 18-29 | 50,6 | 43,0 |
| 30-59 | 50,6 | 43,7 |
Приложение А10. Соотношение жиров и углеводов в диете при разных формах FAOD
| Заболевание (сокращенно) | Диета (доля от общей энрегетической ценности суточного рациона калорийности) |
|---|---|
|
OCTN2, MCAD
ETF-B2+
Мягкая, поздняя, мышечная, асимптотическая формы: CPT2, VLCAD, SCAD, ETF/ETF-DH
HMGSC2, OXCT
|
Нормальная: жиры: 30%;
углеводы: 50-55%;
белки: 10-15%
|
| CACT, CPT2, M/SCHAD, ETF/ETF-DH, MKAT |
Высокоуглеводная, низкожировая:
жиры: 20-25% (включая незаменимые жирные кислоты);
углеводы: 65-75%;
белки: 8-10%.
|
| CPT1, CACT, CPT2, VLCAD, LCHAD, FATP1 |
Высокоуглеводная, низкожировая:
жиры: 25-30% от суточного каллоража (включая: 10% от суточного каллоража ДЦТ, в том числе 3-4% незаменимые жирные кислоты и 15-20% от суточного каллоража - МСТ,);
углеводы: 65-75%;
белки: 8-10%.
|
Приложение А11. Добавки незаменимых жирных кислот при диете с низким содержанием жиров (D-A-CH)
| Возраст | % энергии | ||
|---|---|---|---|
| n–6 линолевая кислота | n–3 ά-линоленовая кислота | Суммарно | |
| от 0 до <4 месяцев | 4,0 | 0,5 | 4,5 |
| от 4 до <12 месяцев | 3,5 | 0,5 | 4,0 |
| от 1 до <4 лет | 3,0 | 0,5 | 3,5 |
| >4 лет | 2,5 | 0,5 | 3,0 |
Приложение А12. Применение углеводов при подозрении на метаболический криз и при интеркуррентных заболеваниях
| возраст | Р-р декстрозы** (%) | Доза мл/кг/ч |
Доза
мл/кг/сут
|
Энергия кг/сут |
|---|---|---|---|---|
| 0-6мес. | 15 | 7,7 | 183 | 110 |
| 6-12мес. | 15 | 7,0 | 168 | 100 |
| 1-3г. | 20 | 4,5 | 110 | 90 |
| 3-6л. | 25 | 3,3 | 80 | 80 |
| 6-12л. | 25 | 2,6 | 65 | 65 |
| или 6-12л. | 30 | 2,25 | 54 | 65 |
| 12-15лет | 30 | 1,8 | 42 | 50 |
| >15лет | 30 | 1,6 | 38 | 45 |
| Добавка глюкозы (мг/кг в минуту) в/в |
Углеводы per os (25% раствор)
|
Дневная дозировка |
|---|---|---|
| 8 (до 2 лет) | Мальтодекстрин | 0,8 г/кг/2ч = 12*3,2 мл/кг в день |
| 6 (до 2-8 лет) | Кукурузный крахмал (сырой) (с 2 лет) | 1,5 г/кг/4ч = 6*6 мл/кг в день |
| 5,5 (старше 8 лет) | 2 г/кг/6ч = 4*8 мл/кг в день |
Приложение А13. Содержание МСТ в специализированных продуктах дополнительного питания, применяемых у пациентов с НОЖК
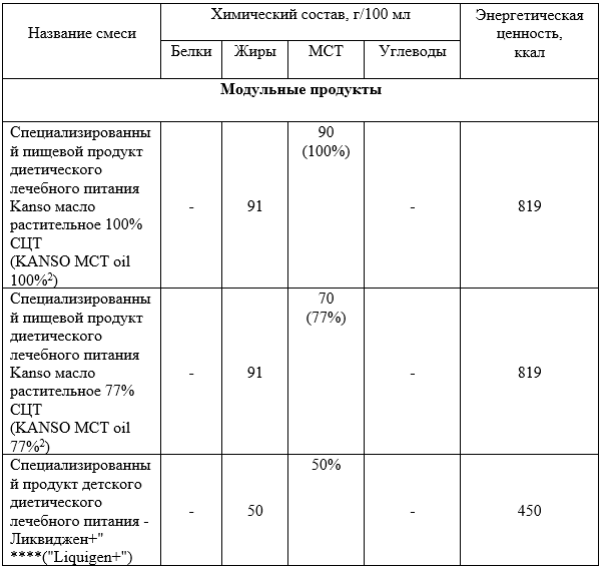
Приложение А14. Диета при недостаточности TFP/ LCHAD (консенсус экспертов)
|
Недостаточность
TFP
|
Возраст | Жиры | Углеводы | Калории |
|---|---|---|---|---|
|
Бессимптомная
или
симптоматическая
|
0–4 месяцев |
Исключение грудного молока или детских смесей, 100% специальной смеси с низким содержанием жиров
· Monogen, SHS
Количество на 100 мл: LCT 0,21 г, MCT 1,89 г + незаменимые жирные кислоты (3,5 г/день)
или
|
Без специальных добавок (только при клинических показаниях) |
Без специальных добавок
(рекомендации D-A-CH)
|
|
после 4 месяцев
(введение в рацион твердой пищи)
|
Снижение содержания жиров до 25–30% от общей калорийностиa; 20–25% жиров за счет MCTb; 3–4% жиров за счет незаменимых жирных кислот
(рекомендации D-A-CH).
Потребление LCT (помимо основных жирных кислот) должно быть максимально низким!
|
Приложение А15. Диета при недостаточности VLCAD (консенсус экспертов)
| Вариант VLCADD | Возраст | Жиры | Углеводы | Калории |
|---|---|---|---|---|
| Бессимптомная (и CK, AST и ALT в пределах нормы) | 0–4 месяцев |
Половина грудное молоко/детская смесь, половина – специальная смесь с низким содержанием жиров
· Monogen, SHS количество на 100 мл: LCT 0,21 г,
MCT 1,89 г
или
|
Без специальных добавок (только при клинических показаниях) |
Без специальных добавок
(рекомендации D-A-CH)
|
|
после 4 месяцев
(введение в рацион твердой пищи)
|
Снижение содержания жиров до 30–40% от общей калорийностиa
(рекомендация D-A-CH);
10–15% жиров за счет MCT
|
|||
| Симптоматичная | 0–4 месяцев |
Исключение грудного молока или детских смесей,
100% специальная смесь с низким содержанием жиров)
· Monogen, SHS
Количество на 100 мл: LCT: 0,21 г,
MCT: 1,89 г + незаменимые жирные кислоты (3,5 г/день)
или
·
|
Без специальных добавок
(только при клинических показаниях)
|
Без специальных добавок
(рекомендации D-A-CH)
|
|
после 4 месяцев
(введение в рацион твердой пищи)
|
Снижение содержания жиров до 25–30(-40) % отобщейототобщейототобщейототобщейототобщейототобщейот общей калорийностиa; 20% жиров за счет MCTb; 3–4% жиров за счет незаменимых жирных кислот (рекомендации D-A-CH) |
MCT – среднецепочечные триглицериды; LCT – длинноцепочечные триглицериды; D-A-CH – рекомендации объединения нутриционистов Германии/Австрии/Швейцарии.
Приложение А16. Натуральные источники незаменимых жирных кислот [105]
| Источник | Количество | Длинноцепочечные жиры (г) | Линолевая кислота (мг) | Линоленовая кислота (мг) | Энергетическая ценность (ккал) |
|---|---|---|---|---|---|
| Льняное масло | 1 мл | 0,9 | 114 | 480 | 8 |
| Рапсовое масло | 1 мл | 0,9 | 183 | 84 | 8 |
| Масло грецкого ореха | 1 мл | 0,9 | 476 | 94 | 8 |
| Сафлоровое (шафрановое) масло | 1 мл | 0,9 | 672 | 0 | 8 |
| Кукурузное масло | 1 мл | 0,9 | 482 | 10 | 8 |
| Соевое масло | 1 мл | 0,9 | 459 | 61 | 8 |
Приложение А17. Состав смесей для диетического питания с пониженным содержанием жира и длинноцепочечных жирых кислот
Предлагается следующая таблица:
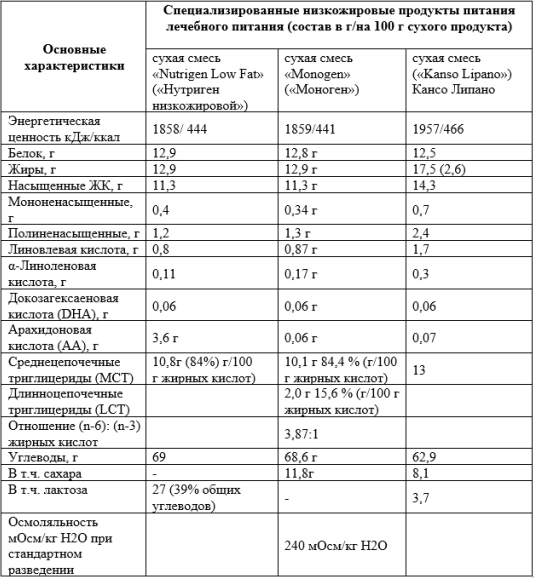
Приложение А18. Пример расчета диеты у пациентов с нарушениями окисления жирных кислот
Пример № 1 Диагноз: недостаточность длинноцепочечной гидроксил-КоА дегидрогеназы (LCHAD)
Возраст 12 дней (период постановки диагноза)
Масса – 3,2кг.
Назначение специализированного продукта «Нутриген Низкожировой»/ «Моноген»
Расчет диеты:
| время | Смесь «Моноген» мл. |
Общ.
Жиры
|
МСТ | ПНЖК | ккал |
|---|---|---|---|---|---|
| 6:00 | 60,0 | 1,32 | 1,08 | 0,13 | 44,76 |
| 9:00 | 60,0 | 1,32 | 1,08 | 0,13 | 44,76 |
| 12:00 | 60,0 | 1,32 | 1,08 | 0,13 | 44,76 |
| 15:00 | 60,0 | 1,32 | 1,08 | 0,13 | 44,76 |
| 18:00 | 60,0 | 1,32 | 1,08 | 0,13 | 44,76 |
| 21:00 | 60,0 | 1,32 | 1,08 | 0,13 | 44,76 |
| 24:00 | 60,0 | 1,32 | 1,08 | 0,13 | 44,76 |
| 03:00 | 60,0 | 1,32 | 1,08 | 0,13 | 44,76 |
| Всего (г) | 10,56 | 8,64 | 1,04 | 358 | |
| Всего % | 26,5 | 21,7 | 2,61 |
Пример №2 Диагноз: недостаточность очень длинноцепочечной ацил-КоА дегидрогеназы (VLCAD) с ранней манифестацией
Возраст 12мес.
Количество общего жира:
Количество МСТ:
В) Максимальные интервалы между кормлениями 4 часа днем, 6 часов ночью (под контролем уровня гликемии), что соответствует 6 приемам пищи.
Калорийность разового кормления примерно составляет 960ккал/6 = 160ккал
Количество общего жира в разовом кормление примерно составляет 27г/6 =4,5г
Количество МСТ в разовом кормление примерно составляет 23г/6 =3,8г
Примерное меню для ребенка 12 мес.
| Прием пищи | продукт | объем | белки |
Общ.
Жиры
|
МСТ | ПНЖК | углеводы | ккал |
|---|---|---|---|---|---|---|---|---|
|
5:30
|
Специализированная
смесь «Нутриген Низкожировой»/ «Моноген»
|
33,6 (г.) 6м.л. | 4,3 | 4,3 | 3,6 | 0,45 | 23,2 | 148 |
| Вода | 180мл | 0 | 0 | 0 | 0 | 0 | 0 | |
|
Liquigen****
ИЛИ
Кансо МСТ масло 100%
|
3 мл
1,5 мл
|
0 | 1,5 | 1,5 | 0 | 0 | 12,5 | |
| 9:30 | Каша кукурузная (сухая) | 25г. | 1,8 | 0,3 | 0 | 0 | 21 | 93,2 |
| Молоко 0,5% 30мл +170 мл воды | 200мл | 0,8 | 0,15 | 0 | 0 | 1,5 | 10,5 | |
|
Liquigen****
ИЛИ
Кансо МСТ масло 100%
|
8мл
4 мл
|
0 | 4 | 4 | 0 | 0 | 33,2 | |
| Пюре из груши | 50г | 0,2 | 0 | 0 | 0 | 5,5 | 22,8 | |
| Масло грецкого ореха | 1мл | 0 | 1 | 0 | 0,7 | 0 | 9 | |
| 13:30 |
Специализированная
смесь «Нутриген Низкожировой»/ «Моноген»
|
33,-35(г.) | 4,3 | 4,3 | 3,6 | 0,45 | 23,2 | 148 |
| Вода | 180мл | 0 | 0 | 0 | 0 | 0 | 0 | |
|
Liquigen****
ИЛИ
Кансо МСТ масло 100%
|
3мл
1,5 мл
|
0 | 1,5 | 1,5 | 0 | 0 | 12,5 | |
| 16:30 | картофель | 100г. | 2,0 | 0 | 0 | 0 | 16 | 72,9 |
| морковь | 30г. | 0,3 | 0 | 0 | 0 | 1,7 | 8 | |
| кабачки | 30г. | 1 | 0 | 0 | 0 | 0,7 | 6,8 | |
| Цветная капуста | 30г. | 0,4 | 0 | 0 | 0 | 1,3 | 7,7 | |
| Филе индейки | 30г. | 5,8 | 0,5 | 0 | 0 | 0 | 27,7 | |
| Liquigen | 8мл | 0 | 4 | 4 | 0 | 0 | 33,2 | |
| Масло грецкого ореха | 1мл | 0 | 1 | 0 | 0,7 | 0 | 9 | |
| 20:00 | Творог обезжиренный 0,2% | 100г. | 16 | 0,1 | 0 | 0 | 1,8 | 72,1 |
| Яблочное пюре | 100г | 0,2 | 0 | 11,3 | 46 | |||
|
Liquigen****
ИЛИ
Кансо МСТ масло 100%
|
8 мл
4 мл
|
0 | 4 | 4 | 0 | 0 | 33,2 | |
| Масло грецкого ореха | 1мл | 0 | 1 | 0 | 0,7 | 0 | 9 | |
|
24:00
|
Специализированная
смесь «Нутриген Низкожировой»/ «Моноген»
|
33-35 (г.) | 4,3 | 4,3 | 3,6 | 0,45 | 23,2 | 148 |
| Вода | 180мл | 0 | 0 | 0 | 0 | 0 | 0 | |
|
Liquigen****
ИЛИ
Кансо МСТ масло 100%
|
3мл
1,5 мл
|
0 | 1,5 | 1,5 | 0 | 0 | 12,5 | |
| итог | всего | 41,4 | 33,45 | 27,3 | 3,45 | 130,5 | 975,8 | |
| На кг массы тела | 3,45 | 2,78 | 2,27 | 0,28 | 10,8 | 81,3 | ||
| % | 16,9 | 30,8 | 25,1 | 3,2 | 53,4 | |||
Пример 3 Диагноз: недостаточность длинноцепочечной гидроксил-КоА дегидрогеназы (LCHAD)
Возраст 4 года
Масса – 16кг.
Расчет диеты исходя из потребности в энергии, а также соотношения жиров:
Рекомендуемая калорийность питания в сутки – 16 х 79,7 = 1275ккал
Количество общего жира:
Количество МСТ:
В) Максимальные интервалы между кормлениями 4 часа днем, до 8 часов ночью, что соответствует 5 приемам пищи.
Калорийность разового кормления примерно составляет 1275 ккал/5 = 255ккал
Количество общего жира в разовом кормлении примерно составляет 35г/5 =7г
Количество МСТ в разовом кормлении примерно составляет 30г/5 =6г
| Прием пищи | продукт | объем | белки |
Общ.
Жиры
|
МСТ | ПНЖК | углеводы | ккал |
|---|---|---|---|---|---|---|---|---|
|
7:00
|
Каша кукурузная | 35г | 2,5 | 0,3 | 0 | 0 | 29,4 | 130 |
| Молоко 0,5% 50мл +200 мл воды | 250мл | 1,4 | 0,25 | 0 | 0 | 2,5 | 17,9 | |
|
Liquigen****
ИЛИ
Кансо МСТ 100%
|
12мл
6 мл
|
0 | 6 | 6 | 0 | 0 | 50 | |
| Фрукты (груша) | 100г. | 0,4 | 0 | 0 | 0 | 11 | 47,4 | |
| мед | 10г | 0 | 0 | 0 | 0 | 8,2 | 32,8 | |
| Масло грецкого ореха | 1мл | 0 | 1 | 0 | 0,7 | 0 | 9 | |
| 11:00 | картофель | 150г | 3 | 0 | 0 | 0 | 24 | 109 |
| брокколи | 30г | 1 | 0 | 0 | 0 | 0,8 | 7,2 | |
| Цветная капуста | 30г | 0,8 | 0 | 0 | 0 | 0,7 | 6 | |
| морковь | 30г | 0,3 | 0 | 0 | 0 | 1,7 | 8 | |
| Индейка (вареная) грудка | 50г. | 9,6 | 0,6 | 0 | 0 | 0,2 | 44,6 | |
| Масло грецкого ореха | 1мл | 0 | 1 | 0 | 0,7 | 0 | 9 | |
|
Liquigen****
ИЛИ
Кансо МСТ 100%
|
14мл
7 мл
|
0 | 8 | 8 | 0 | 0 | 66,4 | |
| 15:00 | Творог обезжиренный 0,2% | 180г | 28 | 0,4 | 0 | 0 | 3,2 | 127,6 |
| банан | 50г | 0,6 | 0 | 0 | 0 | 10 | 43,3 | |
| груша | 50г | 0,2 | 0 | 0 | 0 | 5,5 | 23,7 | |
|
Liquigen****
ИЛИ
Кансо МСТ 100%
|
14мл
7 мл
|
0 | 8 | 8 | 0 | 0 | 66,4 | |
| Масло грецкого ореха | 1мл | 0 | 1 | 0 | 0,7 | 0 | 9 | |
| 19:00 | картофель | 200 | 4 | 0 | 0 | 0 | 32 | 145,8 |
| Томат | 50 | 0,5 | 0 | 0 | 0 | 1,3 | 7,2 | |
| огурец | 50 | 0,3 | 0 | 0 | 0 | 0,9 | 4,8 | |
| Филе индейки | 30г. | 5,8 | 0,4 | 0 | 0 | 0,1 | 27,2 | |
|
Liquigen****
ИЛИ
Кансо МСТ 100%
|
14мл
7 мл
|
0 | 8 | 8 | 0 | 0 | 66,4 | |
| Масло грецкого ореха | 1мл | 0 | 0 | 0 | 0,7 | 0 | 9 | |
| 23:00 | Каша рисовая | 35г. | 2,8 | 0,4 | 0 | 0 | 28 | 126,8 |
| Молоко 0,5% 50мл +200 мл воды | 250мл | 1,4 | 0,25 | 0 | 0 | 2,5 | 17,9 | |
| тыква | 50г. | 0,5 | 0 | 0 | 0 | 2,3 | 12,1 | |
| мед | 10г | 0 | 0 | 0 | 0 | 8,2 | 32,8 | |
|
Liquigen****
ИЛИ
Кансо МСТ 100%
|
14мл
7 мл
|
0 | 8 | 8 | 0 | 0 | 66,4 | |
| итог | всего | 63,1 | 43,6 | 38 | 2,8 | 172,2 | 1324 | |
| На кг массы тела | 3,9 | 2,7 | 2,37 | 0,17 | 10,7 | 79,1 | ||
| % | 19 | 29,6 | 24 | 1,9 | 52 | |||
Приложение A19. Частота проведения обследования у пациентов с нарушениями окисления жирных кислот
| Исследования | При постановке диагноза | Каждые 3 мес | Каждые 6 мес |
Каждый
год
|
По показаниям |
|---|---|---|---|---|---|
| Лабораторные исследования | |||||
| Исследование уровня свободного карнитина и ацилкарнитинов в крови | Х | каждые 1-2 месяца пациентам до года | каждые 3 - 6 месяцев у детей младше 6 лет | каждые 6 -12 месяцев пациентам старше 6 лет | В период и при подозрении на метаболический криз |
| Комплексное определение содержания органических кислот в моче | Х | ||||
| Молекулярно-генетическое исследование | Х | ||||
| Общий (клинический) анализ крови развернутый | Х | Х | Х | ||
| Анализ крови биохимический общетерапевтический | Х | Х | В период и при подозрении на метаболический криз | ||
| Исследование кислотно-основного состояния и газов крови (Исследование уровня буферных веществ в крови, Исследование уровня водородных ионов (рН) крови), уровня глюкозы в крови, электролитов (исследование уровня калия, уровня натрия, уровня ионизированного калиция, уровня хлора в крови) | В период и при подозрении на метаболический криз | ||||
| Общий (клинический) анализ мочи, обнаружение кетоновых тел в моче | Х | Х | Х | Х | |
| Исследования уровня N-терминального фрагмента натрийуретического пропептида мозгового (NT-proBNP) в крови | Х | ||||
| Инструментальные исследования | |||||
| Регистрация электрокардиограммы | Х | Х | |||
| Эхокардиография | Х | Х | |||
| УЗИ органов брюшной полости; объем селезенки, печени; УЗИ почек | Х | Х | Х(при наличии патологии чаще) | ||
| Осмотр глазного дна (офтальмоскопия) | Х | Х | Х (при наличии патологии чаще) | ||
| Электроэнцефалография (ЭЭГ) | Х | Х (при наличии эпилептических приступовчаще) | |||
| Консультации специалистов | |||||
| Консультация врача-генетика | Х | Х | |||
| Консультация врача- педиатра/ врача-терапевта | Х | Х | |||
| Консультация врача- невролога | Х | Х | |||
| Консультация врача-диетолога | Х | ||||
| Консультация врача-гастроэнтеролога | Х | ||||
| Консультация врача-офтальмолога | Х | Х | |||
| Консультация врача- кардиолога, врача-детского кардиолога | Х | Х | |||
| Консультация врача-нефролога | Х | ||||
| Консультация врача-эндокринолога | Х | ||||
| Консультация/консультации медицинского психолога | Х | Х | |||
| Консультация врача-физиотерапевта и врача по лечебной физкультуре | Х | ||||
Приложение А20. Пример «экстренной памятки»
1. Показания к госпитализации:
2. План действия:
Соответствие между возрастом ребенка и дозой декстрозы для энтерального применения при подозрении на метаболический криз.
| возраст | Р-р декстрозы (%) | Доза мл/кг/ч |
Доза
мл/кг/сут
|
Энергия ккал/кг/сут |
|---|---|---|---|---|
| 0-6мес. | 15 | 7,7 | 183 | 110 |
| 6-12мес. | 15 | 7,0 | 168 | 100 |
| 1-3г. | 20 | 4,5 | 110 | 90 |
| 3-6л. | 25 | 3,3 | 80 | 80 |
| 6-12л. | 25 | 2,6 | 65 | 65 |
| Или 6-12л. | 30 | 2,25 | 54 | 65 |
| 12-15лет | 30 | 1,8 | 42 | 50 |
| >15лет | 30 | 1,6 | 38 | 45 |
2.3 При госпитализации в стационар, начать непрерывную инфузию р-ром 10% декстрозы** из расчета: новорожденные и дети до 3-х лет 10-12мг/кг/мин, от 3 до 10 лет 8-10 мг/кг/мин, старше 10 лет 5-8 мг/кг/мин
3. Ведение пациента после неотложной ситуации:
4. Противопоказано:
Приложение А21. Перечень основных лабораторных исследований, которые могут помочь в оказании медицинской помощи при состоянии острой метаболической декомпенсации
| №п/п | Лабораторный тест | Комментарий |
|---|---|---|
| 1 | Исследование кислотно- основного состояния и газов в крови | Оценка степени ацидоза |
| 2 | Измерение уровня глюкозы в крови | Оценка степени гипогликемии |
| 3 | Измерение креатинфосфокиназы в крови | Оценка степени рабдомиолиза |
| 4 |
Исследование уровня альбумина в крови. Исследование уровня общего билирубина в крови
|
Заболевания печени можно увидеть у пациентов с тяжелой метаболической декомпенсацией |
| 5 | Измерение уровня молочной кислоты в крови | Может быть повышен при гиповолемии |
| 6 | Исследование уровня свободного карнитина и ацилкарнитинов в крови | Может использоваться для оценки степени тяжести заболевания |
| 7 | Регистрация электрокардиограммы | Измеряют при нарушении метаболизма LCFA и карнитина при значительно повышенном КФК и признаках кардиомиопатии |
| 8 | Эхокардиография |
Приложение А22. Расшифровка примечаний
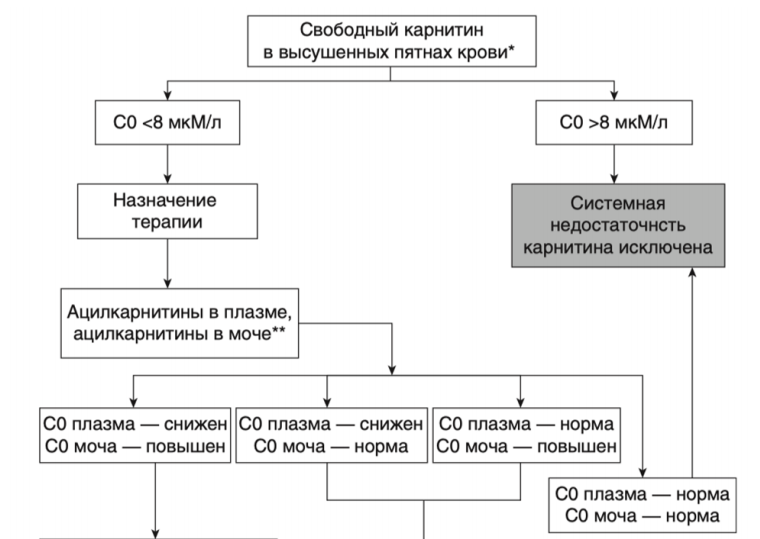
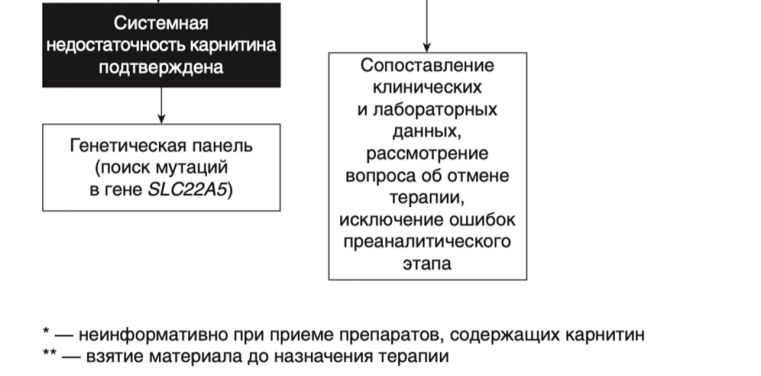
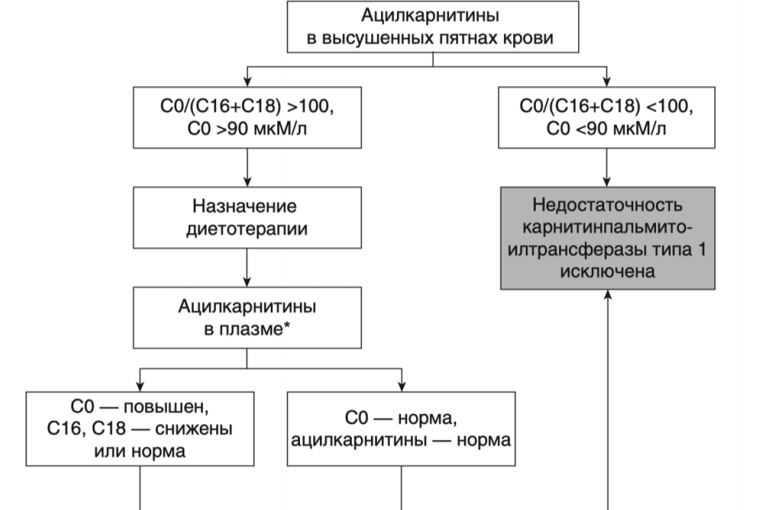
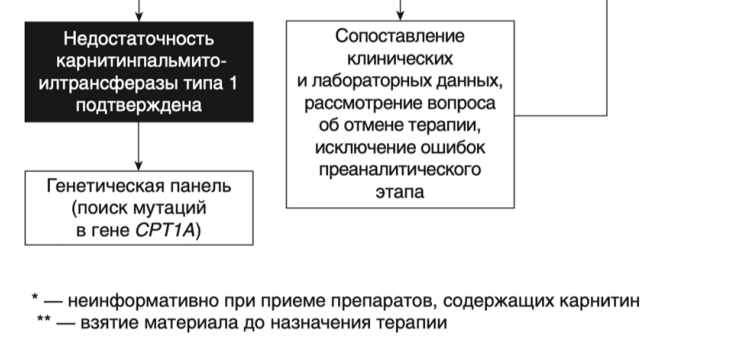
Алгоритм неонатального скрининга карнитинпальмитоилтрансферазы II
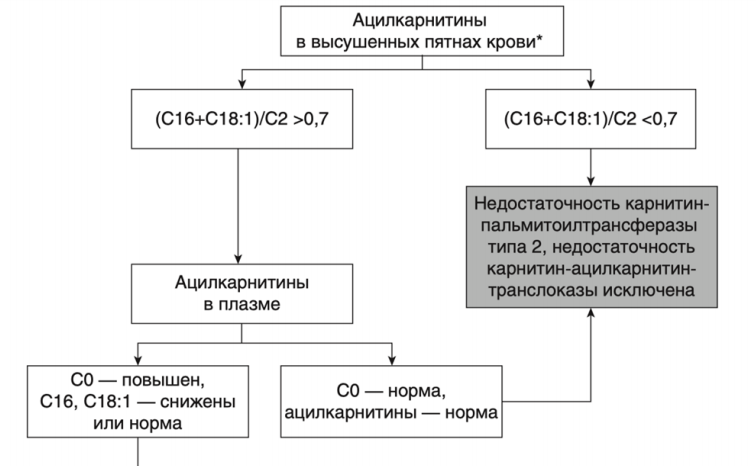

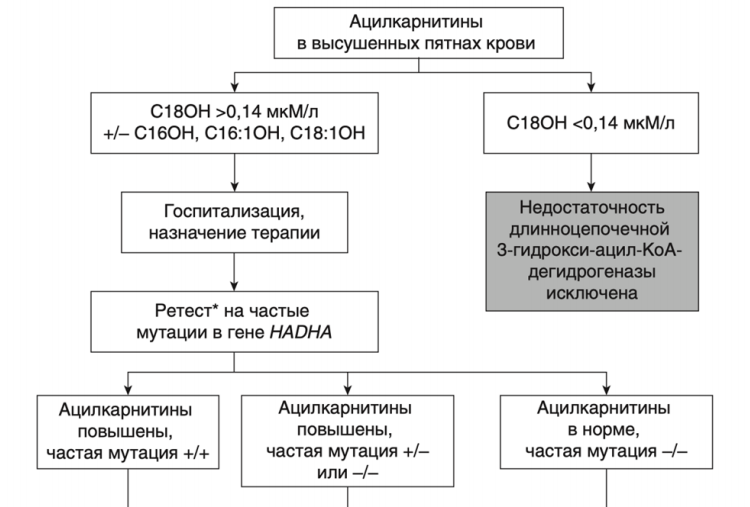
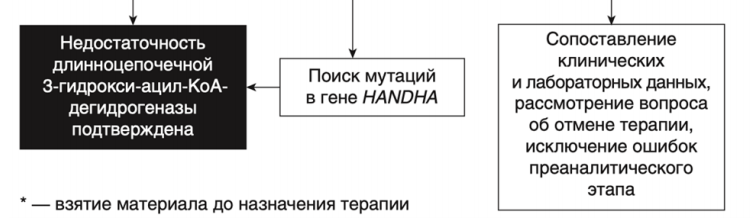
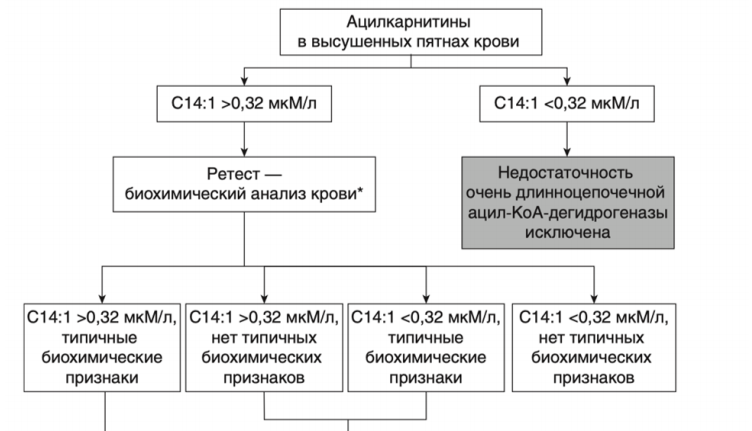
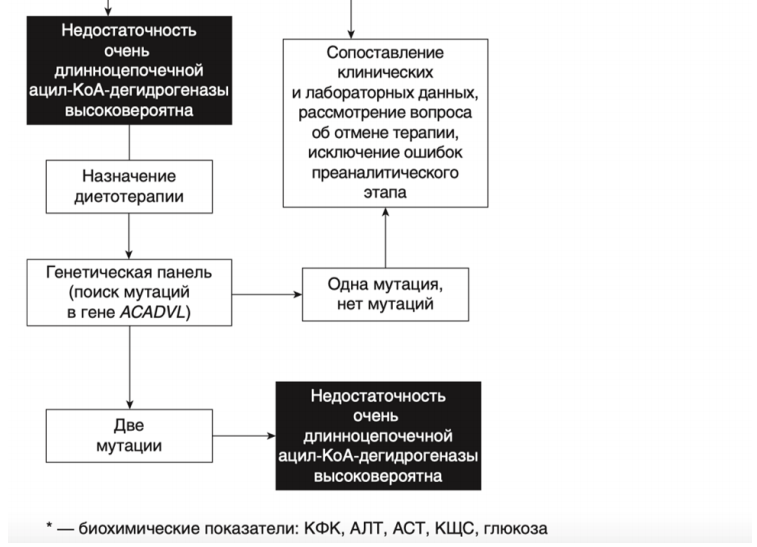
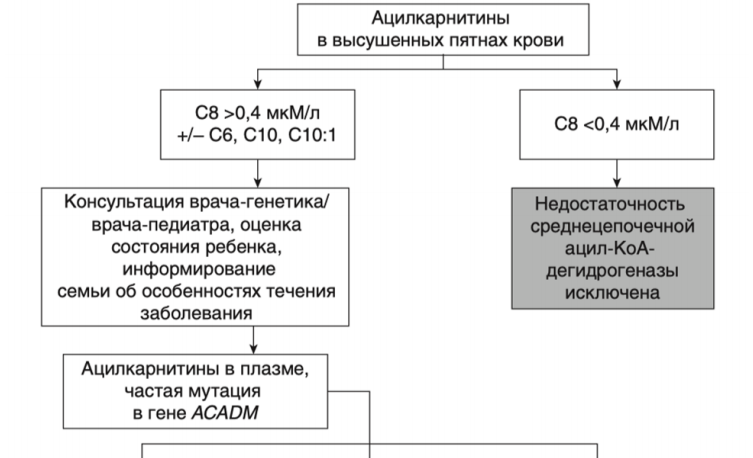
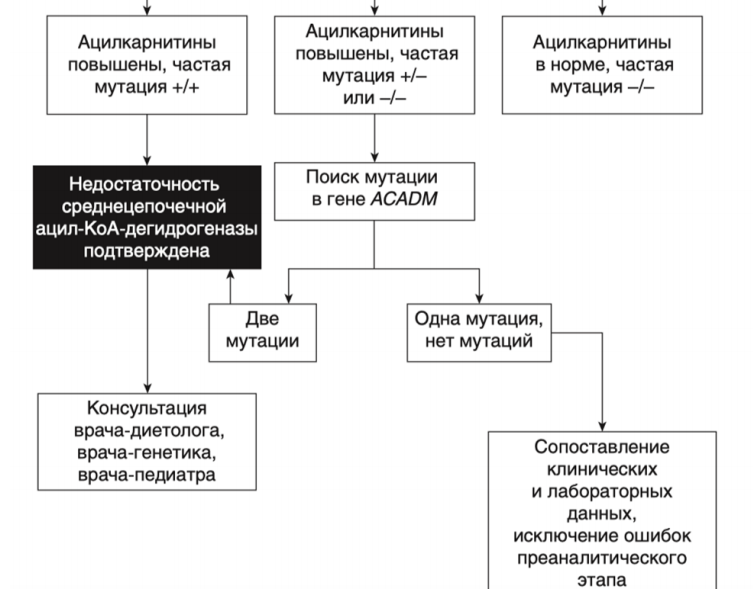
МЕТАБОЛИЗМ И НАРУШЕНИЯ ОБМЕНА ВЕЩЕСТВ
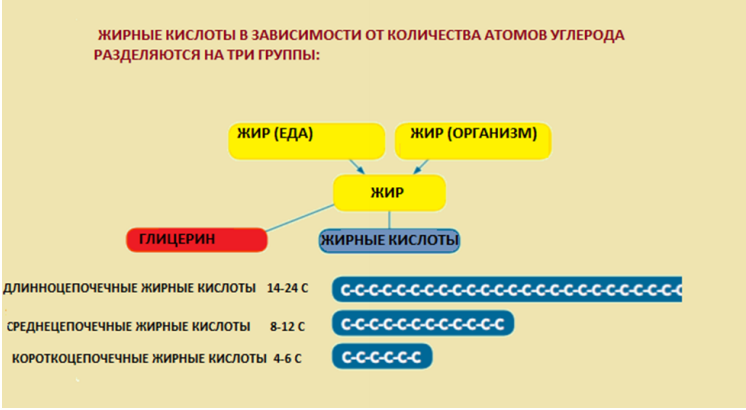
Рис.1. Классификация жирных кислот
β-ОКИСЛЕНИЕ ЖИРНЫХ КИСЛОТ
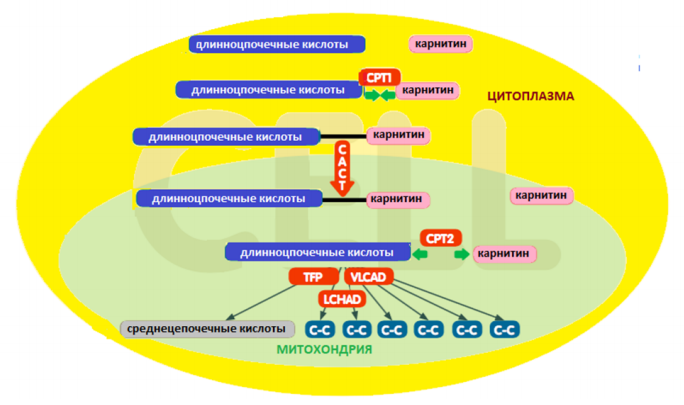 Рис.2. Схема в-окисления жирных кислот.
Рис.2. Схема в-окисления жирных кислот. 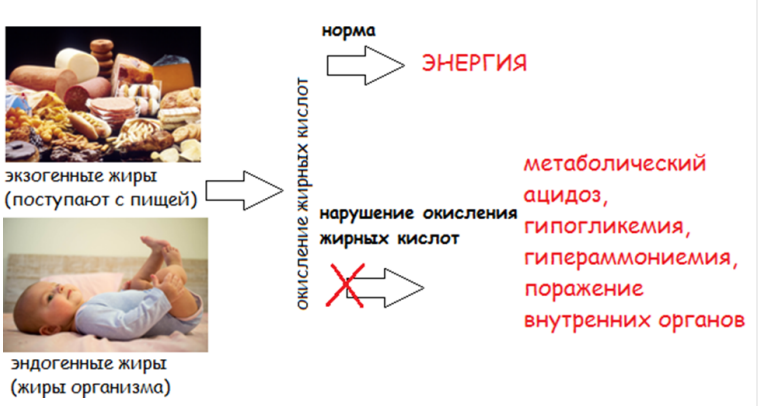
КЛАССИФИКАЦИЯ НАРУШЕНИЙ в-ОКИСЛЕНИЯ ЖИРНЫХ КИСЛОТ И ТРАНСПОРТА КАРНИТИНА
1. Нарушение транспорта жирных кислот
- Первичный дефицит карнитина (ген SLC22A5)
- Клинические проявления: кардиомиопатия, аритмия, сердечная недостаточность, мышечная слабость, поражение печени.
- Карнитин пальмитоилтрансферазы 1 дефицит (ген СРТI)
- Клинические проявления: тяжелое поражение печени, нарушение ее функции, холестаз.
- Карнитин транслоказы дефицит (ген SLC25A20)
- Клинические проявления: тяжелая кардимиопатия, аритмии, поражение печени.
- Карнитин пальмитоилтрансферазы 2 дефицит (ген СРТII)
Клинические проявления: при ранней манифестации (неонатальная форма) кардиомиопатия, поражение печени.
Поздняя манифестация, у взрослых, более мягкая форма, проявляется эпизодами мышечной слабости, трудностями при выполнении физической нагрузки.
2. Нарушение в-окисления жирных кислот
- Короткоцепочечной ацил-КоА дегидрогеназы дефицит (SCAD) (ген ACADS): обычно протекает бессимптомно.
- Среднецепочечной ацил-КоА дегидрогеназы дефицит MCAD (ген АСADM)
- Клинические проявления: может манифестировать в любом возрасте, проявляется быстро прогрессирующим метаболическим кризом после длительного голодания или физической нагрузки. Тошнота, рвота, часто при нормальном уровне глюкозы в крови. При отсутствии терапии может привести к судорогам, коме и остановке сердца.
- Множественный дефицит ацил-КоА дегидрогеназ – глутаровая ацидурия тип II (ген ETFA)
- Клинические проявления очень разнообразны: пороки развития головного мозга, кистозная болезнь почек, гипогликемия, ацидоз, эпилепсия, кардимиопатия.
- Митохондриального трифункционального белка дефицит (МТР) длинноцепочечной гидроксил-КоА дегидрогеназы дефицит (LCHAD) (ген HADHA)
- Клинические проявления: ранняя манифестация, может включать кардиомипатию, нарушение работы печени, мышечную слабость, нейропатию, ретинопатию.
- Очень длинноцепочечной ацил-КоА дегидрогеназы дефицит VLCAD (ген ACADVL)
- Клинические проявления: данное заболевание варьирует от умеренных, слабо выраженных симптомов у одних, до более серьезных проблем у других. Первые симптомы могут проявиться как с рождения, так и уже во взрослой жизни.
Младенческая форма - более тяжелая, протекает как LCHAD, манифестирует между рождением и 4 месяцами, может включать кардиомипатию, нарушение работы печени, мышечную слабость, нейропатию, ретинопатию
- HMG- КоА синтазы дефицит (ген HMGCS2)
- HMG- КоА лиазы дефицит (ген HMGCL)
ГЕНЕТИЧЕСКИЕ ПРИЧИНЫ НАРУШЕНИЯ ОКИСЛЕНИЯ ЖИРНЫХ КИСЛОТ
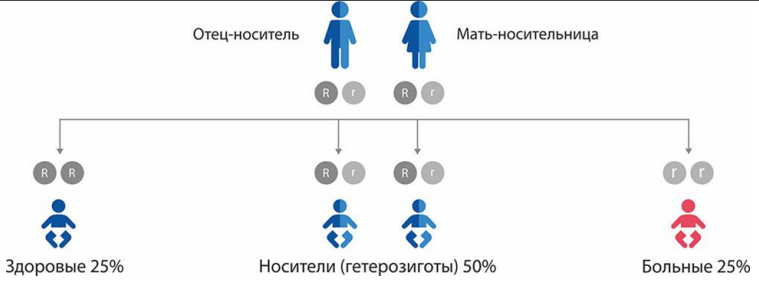
ДИАГНОСТИКА НАРУШЕНИЙ ОКИСЛЕНИЯ ЖИРНЫХ КИСЛОТ
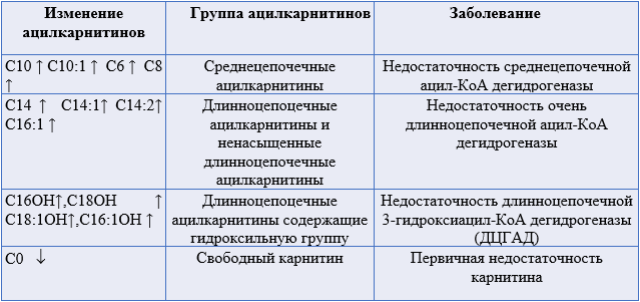
Таблица 1. Соответствие между некоторыми FAOD и изменениями уровней ацилкарнитинов
ДНК-ДИАГНОСТИКА
МОГУТ ЛИ ДРУГИЕ ЧЛЕНЫ СЕМЬИ ЗАБОЛЕТЬ ИЛИ БЫТЬ НОСИТЕЛЯМИ?
ОСОБЕННОСТИ ТЕЧЕНИЯ ЗАБОЛЕВАНИЙ
Для некоторых форм более характерно поражение мышечной системы, для других - сердечной мышцы. Некоторые заболевания протекают преимущественно с поражением печени и с развитием метаболических кризов с гипогликемией.
ОБРАЗ ЖИЗНИ, ЕЖЕДНЕВНАЯ ТЕРАПИЯ И ДИЕТА
Цели:
- Нормальное физическое и интеллектуальное развитие.
- Прибавка в весе и росте, в соответствие с возрастом.
- Предупреждение характерных симптомов, например, мышечной боли, мышечной слабости.
- Предотвращение опасных проявлений, таких как повреждение сердечной мышцы.
- Предупреждение катаболических ситуаций, которые могут привести к метаболическим кризам с гипогликемией и разрушению мышечных клеток.
Для достижения этих целей диетотерапия должна отвечать следующим требованиям:
- Обеспечивать адекватное потребление белка в соответствие с возрастом и потребностью.
- Обеспечивать незаменимыми жирными кислотами, витаминами и минералами.
- покрывать энергозатраты.
- Избегать длительных периодов голодания
- Ограничивать потребление жирных кислот с длинной цепью.
- Соответствовать вкусу, возрасту и потребностям ребенка.
- Подходить для повседневной жизни.
Заболевания, при которых диета не используется:
- Первичный дефицит карнитина
- Короткоцепочечной ацил-КоА дегидрогеназы дефицит (SCAD)
- Среднецепочечной ацил-КоА дегидрогеназы дефицит MCAD
Заболевания, при которых используется диетотерапия:
- Карнитин пальмитоилтрансферазы 1 дефицит (СРТI)
- Карнитин пальмитоилтрансферазы 2 дефицит (СРТII)
- Митохондриального трифункционального белка дефицит (МТР) длинноцепочечной гидроксил-КоА дегидрогеназы дефицит (LCHAD)
- Очень длинноцепочечной ацил-КоА дегидрогеназы дефицит (VLCAD)
- Множественный дефицит ацил-КоА дегидрогеназ
- HMG- КоА синтазы дефицит
- HMG- КоА лиазы дефицит
1. Умеренное ограничение жира в рационе:
- Карнитин пальмитоилтрансферазы 1 дефицит
2. Умеренное или значительное снижение жира в рационе:
- Карнитин пальмитоилтрансферазы 2 дефицит
- Очень длинноцепочечной ацил-КоА дегидрогеназы дефицит VLCAD (бессимптомная форма)
- Множественный дефицит ацил-КоА дегидрогеназ
3. Значительное снижение жира с использованием МСТ (среднецепочечных триглицеридов) +ночные кормления:
- Митохондриального трифункционального белка дефицит (МТР) длинноцепочечной гидроксил-КоА дегидрогеназы дефицит (LCHAD)
- Очень длинноцепочечной ацил-КоА дегидрогеназы дефицит VLCAD
Таблица 2. Максимальный период голода в зависимости от возраста:
| возраст | интервалы между кормлениями |
|---|---|
| Новорожденные | 3 часа днем и ночью |
| <6 месяцев | 4 часа днем и ночью |
| > 6 месяцев-12 месяцев | 4 часа днем, и 6-8 часов ночью |
| > 12 месяцев | 4 часа днем, 10 часов ночью |
Внимание! Если у детей нет проблем со вксармливанием, ночное голодание может быть продлено до 6 часов, начиная с 3-месячного возраста и до 8 часов, начиная с 6-месячного возраста. С осторожностью разрешать голодание более 10 часов даже после первого года лечения. Риск ночной гипогликемии снижается по мере взросления ребенка, и большинство детей после 4 лет, могут переносить периоды без приема пищи 10-12 часов если чувствуют себя хорошо
Таблица 3. Среднесуточные нормы физиологических потребностей в энергии
|
Среднесуточные нормы физиологических потребностей в энергии для детей
согласно FAO/WHO/UNU 2001 (WHO Technical Report Series 17-24okt 2001)
|
||
|---|---|---|
| Возраст (мес.) | Энергия (ккал/кг) муж |
Энергия (ккал/кг)
жен
|
| 0-1 | 113 | 107 |
| 1-2 | 104 | 101 |
| 2-3 | 95 | 94 |
| 3-4 | 82 | 84 |
| 4-5 | 81 | 83 |
| 5-6 | 81 | 82 |
| 6-7 | 79 | 78 |
| 7-8 | 79 | 78 |
| 8-9 | 79 | 78 |
| 9-10 | 80 | 79 |
| 10-11 | 80 | 79 |
| 11-12 | 81 | 79 |
| Возраст (г.) | ||
| 1-2 | 82,4 | 80,1 |
| 2-3 | 83,6 | 80,6 |
| 3-4 | 79,7 | 76,5 |
| 4-5 | 76,8 | 73,9 |
| 5-6 | 74,5 | 71,5 |
| 6-7 | 72,6 | 69,3 |
| 7-8 | 70,5 | 66,7 |
| 8-9 | 68,5 | 63,8 |
| 9-10 | 66,6 | 60,8 |
| 10-11 | 64,6 | 57,8 |
| 11-12 | 62,4 | 54,8 |
| 12-13 | 60,2 | 52,0 |
| 13-14 | 57,9 | 49,3 |
| 14-15 | 55,6 | 47,0 |
| 15-16 | 53,4 | 45,3 |
| 16-17 | 51,6 | 44,4 |
| 17-18 | 50,3 | 44,1 |
| Среднесуточные нормы физиологических потребностей в энергии для людей разных возрастов согласно FAO/WHO/UNU 2007 (WHO Technical Report Series 2007) | ||
|---|---|---|
| Возраст (г.) | Энергия (ккал/кг) муж. | Энергия (ккал/кг) жен |
| Взрослые, ведущие образ жизни средней активности, вес 70кг | ||
| 18-29 | 43,7 | 38,0 |
| 30-59 | 41,8 | 35,3 |
| Взрослые, ведущие образ жизни средней активности, вес 50кг | ||
| 18-29 | 50,6 | 43,0 |
| 30-59 | 50,6 | 43,7 |
Диетотерапия при бессимптомной форме недостаточности очень длинноцепочечной ацил-КоА дегидрогеназы (VLCAD):
- 0-4мес. – 50% грудного молока + 50% специализированное питание с МСТ
- >4мес. – умеренное сокращение жира до 30% энергии от общего калоража, из них 10-15% за счет МСТ, 3-4% за счет незаменимых жирных кислот.
Диетотерапия при недостаточности очень длинноцепочечной ацил-КоА дегидрогеназы (VLCAD) с ранней манифестацией и недостаточности митохондриального трифункционального белка (МТР) недостаточности длинноцепочечной гидроксил-КоА дегидрогеназы (LCHAD):
- 0-4мес – 100% специализированное питание с МСТ
- >4мес. – сокращение жира до 25-30% энергии от общего калоража, 20-25% из них за счет МСТ, 3-4% за счет незаменимых жирных кислот. Снизить потребление длинноцепочечных жирных кислот максимально на сколько возможно.
Для всех типов: избегать катаболических ситуаций!!!
ПРИМЕР РАСЧЕТА ДИЕТЫ
Энергия:
Жиры:
Белки:
Углеводы:
ПОЛИНЕНАСЫЩЕННЫЕ НЕЗАМЕНИМЫЕ ЖИРНЫЕ КИСЛОТЫ (ПНЖК)
Организм всегда использует те жирные кислоты, которые есть в наличии. Именно поэтому целесообразно, включить в рацион ПНЖК класса омега-3 и омега -6 в правильном соотношение.
Омега – 6: линолевая кислота, арахидоновая кислота.
Таблица 4. ПНЖК при низкожировой диете, оптимальное соотношение:
| возраст |
Омега-6
(% от эергии)
|
Омега-3 (%от энергии) | Общая сумма (% от энергии) | Омега-6: Омега-3 | Омега-6 (г/сут) | Омега-3 (г/сут) |
|---|---|---|---|---|---|---|
| 0-<4мес. | 4,0 | 0,5 | 4,5 | 8:1 | 2,0 | 0,25 |
| 4-12мес. | 3,5 | 0,5 | 4,0 | 7:1 | 2,7 | 0,4 |
| 1-<4л. | 3,0 | 0,5 | 3,5 | 6,5:1 | 3,5 | 0,6 |
| >4л. | 2,5 | 0,5 | 3,0 | 5:1 | 5,5 | 1,1 |
Таблица 5. Содержание ПНЖК в растительных маслах:
| масла |
Линоленовая кислота
(омега-6)
|
Альфа-линоленовая кислота
(омега-3)
|
Омега-6:омега-3 |
|---|---|---|---|
| Сафлоровое масло | 75 | 0,47 | 150:1 |
| Тыквенное масло | 49 | 0,5 | 98:1 |
| Льняное масло | 14 | 53 | 0,26:1 |
| Кукурузное масло | 55,5 | 1,0 | 55:1 |
| Оливковое масло | 8,2 | 0,86 | 10:1 |
| Рапсовое масло | 15 | 8,6 | 1,7:1 |
| Кунжутное масло | 42,5 | 0,8 | 53:1 |
| Масло соевых бобов | 53 | 7,7 | 7:1 |
| Масло подсолнечное | 50,1 | 0,2 | 62:1 |
| Масло грецкого ореха | 52,4 | 12,2 | 4:1 |
| Масло зародышей пшеницы | 55,7 | 7,8 | 7:1 |
Докозагексаеновая кислота
КАРНИТИН
В остальных случаях карнитин назначают в небольших дозировках с осторожностью per.os. (доза определяется врачом)
ПРОТИВОПОКАЗАНО внутривенное введение раствора карнитина при нарушении b-окисления жирных кислот!!!
КАКИМ ОБРАЗОМ ПИТАНИЕ РЕБЕНКА МОЖЕТ БЫТЬ АДАПТИРОВАНО К ПИТАНИЮ СЕМЬИ
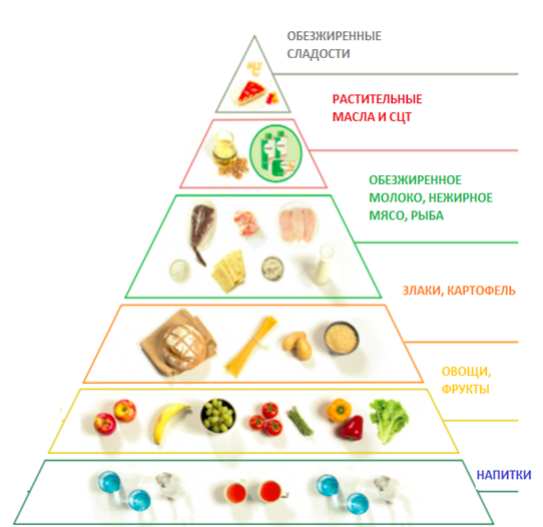
Большая часть продуктов соответствует рекомендациям для здорового, разнообразного питания детей со здоровым метаболизмом. Очень важны напитки без сахара, такие как минеральная вода, фруктовый чай и небольшое количество фруктового сока, смешанного с газированной водой. В приоритете фрукты и овощи. В их составе практически не содержится жиров, поэтому они могут употребляться в большом количестве. Рекомендуется, как и всем, съедать 5 порций фруктов и овощей в день.
Масла и твердые жиры должны быть заменены МСТ и незаменимыми жирными кислотами в правильном соотношении.
МЕТАБОЛИЧЕСКИЙ КРИЗ
Что может спровоцировать метаболический криз:
- длительное голодание
- инфекция
- тяжелая физическая и эмоциональная нагрузка
Признаки гипогликемии (снижение уровня сахара в крови):
- Слабость
- Дрожь
- Головокружение
- Липкий холодный пот.
Первые симптомы метаболического криза:
В КАКИХ СЛУЧАЯХ НУЖНО НЕМЕДЛЕННО ОБРАТИТЬСЯ ЗА МЕДИЦИНСКОЙ ПОМОЩЬЮ
- Плохой аппетит
- Выраженная слабость или чрезмерная сонливость
- Рвота
- Диарея
- Лихорадка
- Постоянные мышечные боли, слабость или красновато-коричневый цвет мочи.
ЧТО ДЕЛАТЬ ЕСЛИ ВЫ ЗАМЕТИЛИ У РЕБЕНКА ЭТИ СИМПТОМЫ?
ТЕРАПИЯ
- Избегать голодания
- Сокращение в рационе жирных кислот с длинной цепью
- Обогащение пищи среднецепочечными жирными кислотами
- Предотвратить гипогликемию
- Дополнительная метаболическая поддержка при развитии инфекционных заболеваний
- Добавки карнитина (см. раздел карнитин)
ФИЗИЧЕСКАЯ НАГРУЗКА
Поэтому если вы знаете, что собираетесь потратить больше энергии чем обычно, вы должны быть к этому готовы.
Специализированное питание
Таблица 7. Состав специализированных низкожировых смесей для питания пациентов при FAOD, начиная с рождения (содержание ингредиентов на 100 г сухого продукта) *
| Основные характеристики |
Специализированные низкожировые продукты питания лечебного питания (состав в г/на 100 г сухого продукта)
|
||
|---|---|---|---|
| сухая смесь «Nutrigen Low Fat» («Нутриген низкожировой») | сухая смесь «Monogen» («Моноген») |
сухая смесь («Kanso Lipano»)
Кансо Липано
|
|
| Энергетическая ценность кДж/ккал | 1858/ 444 | 1859/441 | 1957/466 |
| Белок, г | 12,9 | 12,8 г | 12,5 |
| Жиры, г | 12,9 | 12,9 г | 17,5 (2,6) |
| Насыщенные ЖК, г | 11,3 | 11,3 г | 14,3 |
| Мононенасыщенные, г | 0,4 | 0,34 г | 0,7 |
| Полиненасыщенные, г | 1,2 | 1,3 г | 2,4 |
| Линовлевая кислота, г | 0,8 | 0,87 г | 1,7 |
| α-Линоленовая кислота, г | 0,11 | 0,17 г | 0,3 |
| Докозагексаеновая кислота (DHA), г | 0,06 | 0,06 г | 0,06 |
| Арахидоновая кислота (АА), г | 3,6 г | 0,06 г | 0,07 |
| Среднецепочечные триглицериды (MCT) |
10,8г (84%) г/100
г жирных кислот)
|
10,1 г 84,4 % (г/100 г жирных кислот) | 13 |
| Длинноцепочечные триглицериды (LCT) | 2,0 г 15,6 % (г/100 г жирных кислот) | ||
| Отношение (n-6): (n-3) жирных кислот | 3,87:1 | ||
| Углеводы, г | 69 | 68,6 г | 62,9 |
| В т.ч. сахара | - | 11,8г | 8,1 |
| В т.ч. лактоза | 27 (39% общих углеводов) | - | 3,7 |
| Осмоляльность мОсм/кг H2O при стандартном разведении | 240 мОсм/кг H2O | ||
Таблица 8. Состав специализированных жировых смесей для питания пациентов при FAOD, начиная с рождения (содержание ингредиентов на 100 г сухого продукта) **
| Спец. продукт | Общие жиры % | Из них МСТ % |
|---|---|---|
| Ликвиджен+" ****("Liquigen+") ( | 50 | 45,4 |
| Kanso MCT Oil | 100 | 100 |
ДИСПАНСЕРИЗАЦИЯ
Таблица 8. Регулярность различных исследований при FAOD
| Исследование | интервал |
|---|---|
| ТМС | 1 раз в 3 мес и по показаниям |
| КЩС + лактат | 1 раз в 3 мес и по показаниям |
| Кровь на аммоний | 1 раз в 3 мес и по показаниям |
| Биохимический анализ крови (КФК, АЛТ, АСТ, ЛДГобщий белок, альбумин, глюкоза, холестерин, триглицериды калий, кальций | 1 раз в 3 мес и по показаниям |
| ЭКГ | 1 раз в 6 мес и по показаниям |
| ЭХО-КГ | 1 раз в 6мес и по показаниям |
| ЭЭГ | 1 раз в 6 мес и по показаниям |
| УЗИ органов брюшной полости и почек | 1 раз в 6мес. и по показаниям |
ТЕЧЕНИЕ БЕРЕМЕННОСТИ ПРИ НАРУШЕНИИ В-ОКИСЛЕНИЯ ЖИРНЫХ КИСЛОТ У ПЛОДА
ПРОГНОЗ ПРИ СВОЕВРЕМЕННОЙ ПОСТАНОВКИ ДИАГНОЗА
ДОПОЛНИТЕЛЬНАЯ ИНФОРМАЦИЯ
СОКРАЩЕНИЯ И ПОНЯТИЯ
- СРТ1 - Карнитин пальмитоилтрансферазы 1 дефицит
- СРТ2 - Карнитин пальмитоилтрансферазы 2 дефицит
- SCAD - Короткоцепочечной ацил-КоА дегидрогеназы дефицит
- MCAD - Среднецепочечной ацил-КоА дегидрогеназы дефицит
- МТР - Митохондриального трифункционального белка дефицит
- LCHAD - длинноцепочечной гидроксил-КоА дегидрогеназы дефицит
- VLCAD - Очень длинноцепочечной ацил-КоА дегидрогеназы дефицит
- СЦТ (МСТ) – среднецепочечные жирные кислоты (триглицериды)
- ДЦТ –длинноцепочечные жирные кислоты (триглицериды)
- ПНЖК – полиненасыщенные незаменимые жирные кислоты
- АЦИЛКАРНИТИНЫ - жирные кислоты, связанные с карнитином
- КАРДИОМИОПАТИЯ – заболевание сердца, при которых сердечная мышца структурно и функционально изменена
- ГИПОТОНИЯ – мышечная слабость
- ГИПОГЛИКЕМИЯ - снижение уровня сахара в крови
- РАБДОМИОЛИЗ – разрушение клеток мышечной ткани
- МЕТАБОЛИЧЕСКИЙ АЦИДОЗ - нарушение кислотно-основного состояния, проявляющееся низкими значениями рН крови и низкой концентрацией бикарбоната в крови
- ГИПОГЛИКЕМИЯ – снижение уровня сахара в крови
- ГИПЕРАММОНИЕМИЯ – повышение уровня аммония в крови
- НЕЙРОПАТИЯ - поражение нерва, при котором происходит нарушение его целостности или целостности его оболочки.
- РЕТИНОПАТИЯ - поражение сетчатки
- КАТАБОЛИЗМ – процесс распада сложных веществ.
Приложение Г1-ГN. Шкалы оценки, вопросники и другие оценочные инструменты состояния пациента, приведенные в клинических рекомендациях
Не применимо.
Прикреплённые файлы
Внимание!
- Занимаясь самолечением, вы можете нанести непоправимый вред своему здоровью.
- Информация, размещенная на сайте MedElement и в мобильных приложениях "MedElement (МедЭлемент)", "Lekar Pro", "Dariger Pro", "Заболевания: справочник терапевта", не может и не должна заменять очную консультацию врача. Обязательно обращайтесь в медицинские учреждения при наличии каких-либо заболеваний или беспокоящих вас симптомов.
- Выбор лекарственных средств и их дозировки, должен быть оговорен со специалистом. Только врач может назначить нужное лекарство и его дозировку с учетом заболевания и состояния организма больного.
- Сайт MedElement и мобильные приложения "MedElement (МедЭлемент)", "Lekar Pro", "Dariger Pro", "Заболевания: справочник терапевта" являются исключительно информационно-справочными ресурсами. Информация, размещенная на данном сайте, не должна использоваться для самовольного изменения предписаний врача.
- Редакция MedElement не несет ответственности за какой-либо ущерб здоровью или материальный ущерб, возникший в результате использования данного сайта.